pKa prediction from ab initio calculations
Acidity is one of the most important properties of many chemically active compounds. A compound behaves as an acid if it shows a tendency to transfer one or more hydrogen ions (H+, or protons) to an acceptor species. In the context of biochemical processes in living organisms, often the acceptor species are water molecules. Dissolving an acidic molecule in water will increase the concentration of protons in solution. Vice versa, basic molecules accept protons from water, therefore reducing the H+ concentration.
Weak acids and bases: the pKa
The chemical activity of biological molecules, metabolites and drugs is crucially related to their ability to exchange protons. The biological function of many molecules relies on their ability to carefully regulate how protons are exchanged with other chemical species. The ability of a weak acid to dissociate in water (i.e. donate protons) is measured by its pKa, which is the pH at which half of the molecules in solution are ionised (their proton has dissociated), while the rest of the molecules remain unionised (undissociated).

The pKa of a molecule can be measured experimentally, alternatively, it can be predicted using theoretical models. Knowing a drug’s pKa helps to predict its activity, distribution, metabolism, excretion and toxicity (ADMET) profile without having to synthesise it in the laboratory. In principle, in silico prediction approaches can drastically speed up the screening of large numbers of potential new drugs. Paul Popelier, Beth Caine and their collaborators have developed a simple and efficient approach to pKa prediction, which uses information concerning the 3D structure of one protonation state of a molecule to model its behaviour as a weak acid or base.
Molecular structure and acidity
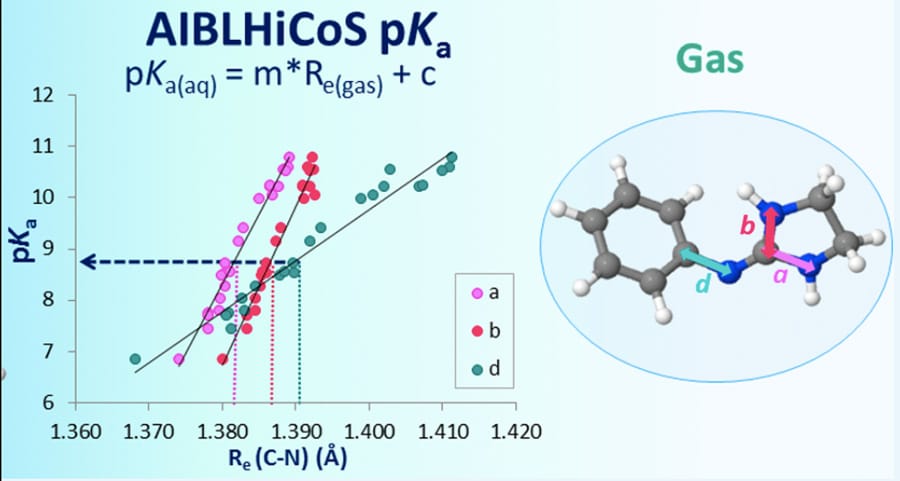
The relative acidity of compounds with similar chemical structures is determined by the electronic properties of other atoms that are present within the molecule (substituent groups). For instance, when a weak acid releases a proton, a negative charge is left behind and it is said to be ionised. The extent to which this charge is redistributed and stabilised throughout the molecule dictates the relative propensity of the compound towards ionisation. The Popelier group have shown that for a series of molecules with similar structures and quantum-chemical properties (electronic congeners), specific bond distances are strongly correlated to pKa values. Consequently, after a model has been calibrated for a series of congeners using experimental data, the only information required to compute pKa values for new compounds is bonding distances for a stable molecular geometry. Crucially, this criterion applies equally well to existing molecules and to new (never synthesised) species, provided a sufficient degree of structural similarity can be identified among them.

Ab initio Bond Lengths pKa prediction
Stable, and therefore commonly observed molecular geometries can be determined using quantum-chemical calculations and a procedure called geometry optimisation. One of the most powerful and commonly used approaches to carry out geometry optimisations (and, in general, electronic structure calculations) is known as density-functional theory (DFT).
Knowing a drug’s pKa makes it possible to predict its activity, distribution, metabolism, excretion and toxicity.
Prof Popelier and his collaborators have systematically studied the application of DFT to the calculation of pKas based exclusively on structural descriptors (equilibrium bond lengths) for several classes of molecules. The team have developed a robust workflow that provides accurate estimates of pKas using regression modelling. Their method, called Ab Initio Bond Lengths-pKa (AIBL-pKa) has been shown to be extremely reliable: not only can it be used to predict pKas of existing or new molecules very accurately, it can also be applied to revise and indeed correct experimental estimates of pKas when these are affected by measurement errors or inaccuracies.
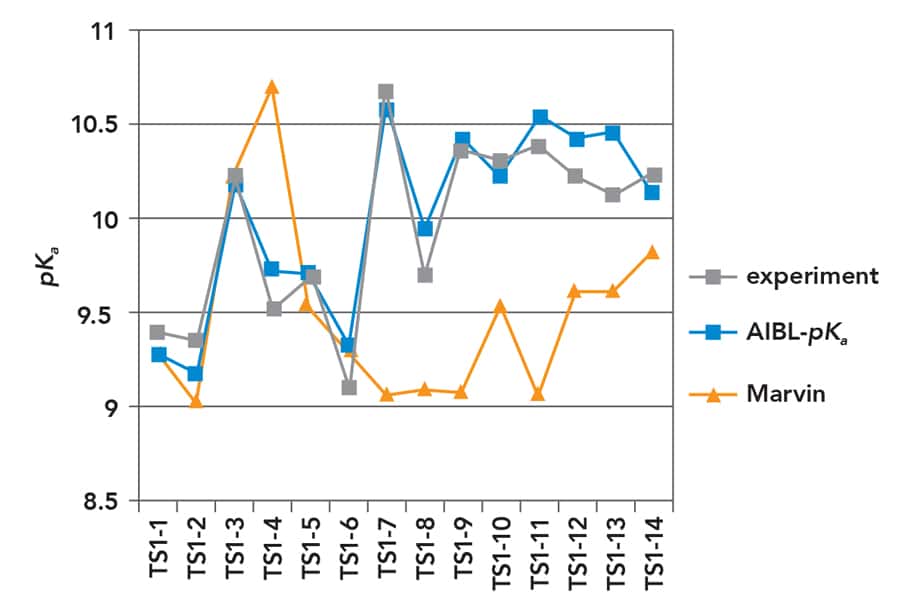
Correlation of pKa and bond lengths
The AIBL-pKa method has been applied successfully to several classes of organic molecules, some of which constitute the building blocks of more complex and biologically important compounds. For instance, in a study of 171 phenol-based molecules (chemical compounds containing an acidic –OH functional group linked to a conjugated phenyl ring), AIBL-pKa has been shown to predict pKas with an accuracy of less than 0.5 logarithmic units compared to experiments. A detailed statistical analysis of potential correlations between molecular structure and pKa has also indicated the existence of a very strong correlation between a single chemical bond length in the molecule (calculated using DFT) and the acidity of the molecule. This is a striking and far reaching result, which shows that pKa values can be estimated to very good accuracy using a single molecular reactivity index (a bond length). Furthermore, this study has demonstrated that the best accuracy in pKa predictions can be achieved by splitting sets of analogue molecules into high correlation subsets (HCSs), which group together molecules with similar structural features, particularly in the close vicinity of the functional group that releases the proton.
The team have developed a robust framework that provides accurate estimates of pKa.
A general approach to pKa calculation
Other instances of where AIBL has been successful include: benzoic acids and anilines, carboxylic acids, amidine and guanidine-based compounds, primary and secondary sulfonamides and a variety of carbon acids. In all cases, these studies have shown the existence of statistically significant bond length/pKa correlations, along with the appearance of HCSs within groups of chemical analogues. The AIBL-pKa method has also been highly successful in cases for which other methods for pKa prediction have been shown to be unreliable or difficult to apply, including molecules containing more than one acidic functional group and systems that exhibit tautomerism, i.e. the coexistence of two rapidly interconverting chemical structures for the same molecule. Tautomerism is observed in several classes of organic and biological compounds, including amino acids (the fundamental constituents of proteins) and nucleic acids (the building blocks of DNA).

Novel application of the AIBL-pKa approach
Empirical predictors need experimental data to train their model(s). For regions of chemical space that are not properly represented in the training set, the accuracy of these methods rapidly deteriorates. A recent study carried out in collaboration with Lhasa Ltd. has shown that the AIBL approach can provide a solution to this problem. Using AIBL-pKa models constructed for variants of carbon acids (a region of chemical space where Lhasa’s empirical-based model performs least well), Professor Popelier and co-workers have constructed hypothetical compounds and predicted their pKa values. These hypothetical compounds have been purposefully built to increase the diversity of atom types found in the carbon acids subset of the training set. Excitingly, their results indicate that the addition of such species to the training set both enhances the accuracy of Lhasa’s predictive tool and makes it and more widely applicable. The current work of Prof Popelier and his collaborators aims to expand further and document the reliability and accuracy of the AIBL-pKa method to systems of increasing complexity. Further work will explore how the approach can be applied to monoterpenes, and thus aid the rational design of new monoterpene synthase enzymes.
Personal Response
One of the most striking findings of your work has been that a complex chemical phenomenon, the dissociation of a weak acid in water, can be rationalised using a simple criterion based on easily calculated structural information on one protonation state. What are the prospects of the AIBL-pKa method in drug discovery, and are there any challenges to its current applicability or potential improvements that it could benefit from?
A caveat of AIBL is that it requires calibration, and each linear model has a defined domain of applicability that is restricted by the availability of experimental data. Furthermore, quantum chemical calculations are more time-consuming than other methods, such as Lhasa’s approach. Their approach uses 2D molecular fingerprints to define a molecular structure (no quantum chemical input features) and returns predicted values in a matter of seconds. As we have mentioned, one exciting example of AIBL’s potential is feeding a fast empirical model with theoretical, yet highly accurate data. This has already been shown to improve accuracy for Lhasa’s approach with the addition of less than 150 hypothetical compounds, whilst still providing the user with an answer on a very short timescale.