Tracking how SARS-CoV-2 mutates around the world during COVID-19 pandemic
Coronaviruses are a family of viruses which cause respiratory infections, with symptoms ranging from very mild (or asymptomatic) to lethal. SARS-CoV-2 (Severe Acute Respiratory Syndrome CoronaVirus 2) is the strain of coronavirus responsible for the COVID-19 pandemic. As of early November it has caused approximately 52 million cases and 1.3 million deaths.
A global effort to amass information about the virus has resulted in the SARS-CoV-2 genome being sequenced thousands of times all across the world and over the duration of the pandemic. This huge data set provides a unique opportunity to track the evolution of the virus as it mutates and spreads. Information resulting from analysis of this data could help with efforts to control and treat the disease, both in terms of developing medications and vaccinations and in terms of government policies to limit spread by reducing contact.

Genomic analyses
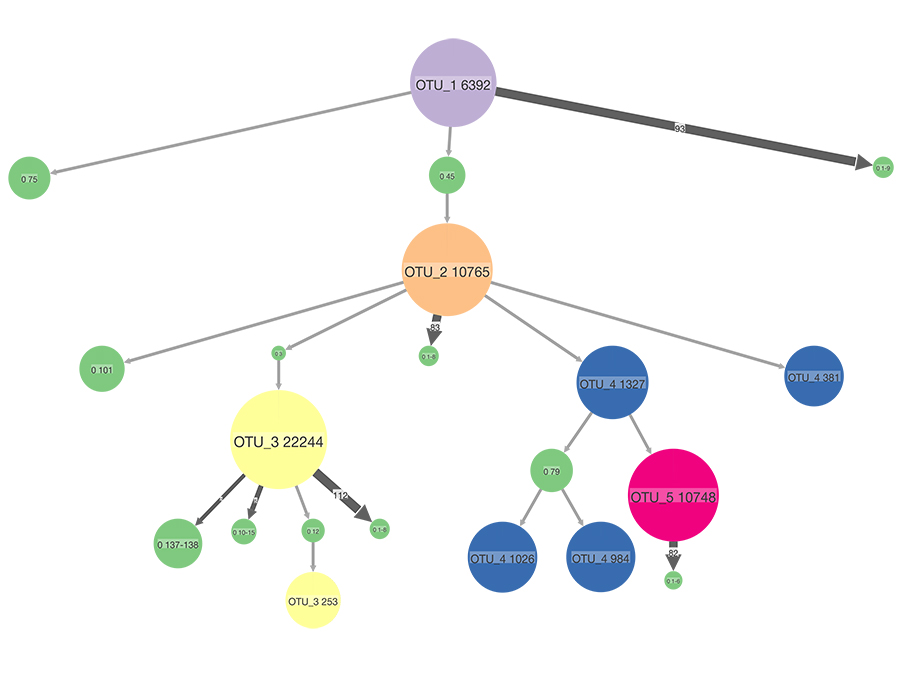
Dr Santiago Justo Arévalo and his team analysed the 171,461 SARS-CoV-2 genomes available at the end of November and found nine frequently occurring mutations. They then used a statistical model to create a tree illustrating the most likely evolution of the virus. The model showed five distinct groups of SARS-CoV-2 genomes that can be characterised by the presence or absence of the nine mutations. These groups are known as haplotypes. A haplotype is a set of genetic material that is usually all inherited together, in this case a distinct set of genes that all copies of the virus will contain when it is replicated inside a cell (viruses reproduce by entering a host cell and forcing the cell to produce copies of the virus). The research team refers to the haplotypes as OTU_1 to OTU_5 (operational taxonomic unit – a functional term used to designate a group of closely related individuals).
OTU_1 is the ancestor haplotype, containing none of the nine mutations identified. It was the first SARS-CoV-2 genome identified in humans, in Asia during December 2019. OTU_2 was first sequenced in January of 2020, and contains four mutations (from the nine identified) that are not present in OTU_1. Around the same time OTU_3 was also sequenced, containing the four mutations found in OTU_2 plus three more. OTU_4 is also descended from OTU_2 and so contains those four mutations plus one additional mutation (but does not contain the three unique to OTU_3). OTU_5 is descended from OTU_4, and so contains the four mutations found in OTU_2, the additional one found in OTU_4, and one further mutation unique to OTU_5.


Estimating OTU frequencies
A problem with analysing the global frequencies of OTUs results from different countries and continents being able to sequence different numbers of genomes. To solve this problem Dr Arévalo and his team applied a statistical process called ‘normalization’. This works by extrapolating how common a haplotype is in the sequenced genomes of one area to the number of cases of the disease in that area. For example, if a country sequenced only a few genomes but all of the sequenced genomes were from one haplotype, you could assume that all of the cases of SARS-CoV-2 (sequenced or not) in that country were also from that haplotype. Then the frequency in terms of the number of cases of that haplotype can be used to give a better representation of that OTU’s global importance.
Dr Arévalo and his team analysed the 171,461 SARS-CoV-2 genomes available at the end of November and found nine frequently occurring mutations.
Temporal distribution
The research team used the data available at the end of November 2020 to track how the OTUs spread over the first eleven months of the pandemic. In January and February, OTU_1 spread from Asia to North America and Europe and was by far the most common haplotype. OTU_2 to OTU_5 were also present but in very low numbers, until March when the frequencies began to even out amongst the five groups. By April, OTU_1 was no longer the most common haplotype. OTU_3 was then the most prevalent globally, but OTU_2 was found frequently in Africa, and OTU_5 had established itself as the most frequent haplotype in North America. OTU_3 increased in frequency and was the most common haplotype worldwide from May to August. OTU_2 and OTU_5 were present in intermediate frequencies, and OTU_1 and OTU_4 were the least commonly reported haplotypes.
However, in September OTU_3 began to decrease in frequency and OTU_2 began to increase. South America was not included in analyses from September onwards and only North America and Europe were considered for November. Because In October and November OTU_2 was the most common globally due to the absence of information of other continents where OTU_3 was still the most common. In October OTU_2 became more common than OTU_3, which continued to decline through November. OTU_1 continued decreasing in occurrence, and by November was rarely reported. OTU_4 and OTU_5 were present in low frequencies, but OTU_4 was slightly more common in August and September than in other months, and OTU_5 was more common in November.
Overall, these results show that while OTU_1 spread the SARS-CoV-2 virus worldwide, it was then outcompeted by later haplotypes with new, potentially advantageous, mutations. OTU_3 contains three mutations unique to this haplotype, which could be the reason why OTU_3 became the most frequent haplotype early on. OTU_3 was later replaced by OTU_2 which doesn’t contain those unique mutations, however due to too few genomes being sequenced South America was not included in analyses from September onwards and only North America and Europe were considered for November. Dr Arévalo and his team were able to break the analysis down further and look at how the haplotypes changed in frequency within continents.
Geographic distribution
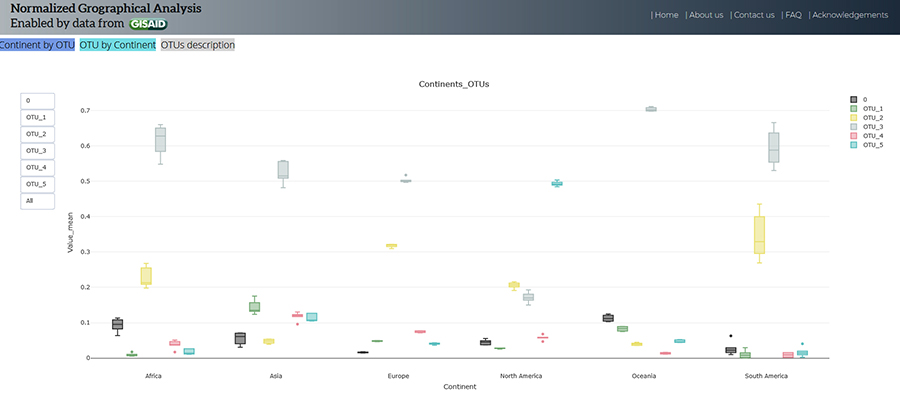
Dr Arévalo and his team found that all continents had cases of all OTUs, but the frequencies of OTUs varied between continents. OTU_3 was the most prevalent haplotype in Asia, Africa, Oceania (Australia and many South Pacific islands) and South America. In North America OTU_5 was found to be most common. In Europe OTU_3 was the most prominent until September when OTU_2 became most common. The research team suggests that these differences could be due to policies put in place by governments to reduce the spread of the disease, or could be the result of mutations beneficial to the virus.

Mutations of interest
A lot of interest lies in the mutation to the spike protein, because this protein allows the process of the virus to fuse to the host cell to begin and is therefore a common target for vaccines. This mutation first appears in OTU_2 and it is suggested it may make for a more infectious virus. However, this mutation is present in OTU_2-5, and so it could be that other mutations are the reason for the particular success of OTU_3, and in North America OTU_5.
The three mutations unique to OTU_3 are in the N protein. The N protein is involved in binding and stabilising the viral genome and in many processes essential for viral replication, so while it is unclear what effect the mutations might have they could provide an advantage to the virus. The N protein of SARS-CoV-2 is another potential target for vaccines.
OTU_5 has one unique mutation in a protein called Nsp2. The function of Nsp2 in SARS-CoV-2 is still unclear but there is a possibility it is involved in modulating host cell processes to allow for viral replication. It is unknown whether the mutation in Nsp2 unique to OTU_5 could be beneficial for the virus and thus be the reason for its high prevalence in North America.
OTUs and patient characteristics
The research team also considered whether there might be any relationships between OTUs and patient age, gender or severity of illness. They found that there was no significant difference in the occurrence of the different haplotypes across gender or age categories, or in the severity of patient symptoms. However, there is a severe shortage of available data for these analyses, and in the case of severity of symptoms what information was available was disorganised and often uninformative.
HaploFinder URP is a tool available to all researchers, where they can upload a sequenced SARS-CoV-2 genome and find out which haplotype it belongs to.
HaploFinder URP
While SARS-CoV-2 is considered a slowly-mutating virus, further mutations will have occurred past the point of these analyses and will continue to occur, and therefore new haplotypes will develop. Dr Arévalo and his team are continuing to monitor the evolution of the virus and the frequencies of global mutations via a webpage they have created called HaploFinder URP.
HaploFinder URP (sarscov2haplofinder.urp.edu.pe) is a tool available to all researchers, where they can upload a sequenced SARS-CoV-2 genome and find out which haplotype it belongs to. The research team provides the results from their genomic analyses on the webpage, and will continue to update this information as new mutations and haplotypes emerge and are identified.

Personal Response
Do you think the results of the frequency analyses would have been different if lockdown policies hadn’t been implemented?
<> The spreading of the mutations that define those five major haplotypes occurred before lockdown policies were completely implemented all around the world. Thus, as you can see in our paper ‘Analysis of the Dynamics and Distribution of SARS-CoV-2 Mutations and its Possible Structural and Functional Implication’, the high-frequency mutations defined here (almost the same that define our five major haplotypes [OTU_1 to OTU_5]) are in all six continents analysed. However, another set of mutations (that appear after lockdown policies) are restricted to some continents. This observation offers evidence that lockdown policies (especially those that avoid intercontinental travel) satisfactorily maintain mutations that appear in one continent are restricted to that continent. If intercontinental travel is reinstated (as is already being observed in some cases), those mutations will begin to spread all around the world and the second round of haplotype competition between continents will begin, if it has not started yet…