New hope for muscular dystrophy treatment
Affecting just over one in a thousand people, muscular dystrophy (MD) is the most widespread lethal genetic disease of humans. Causing muscle weakness and wasting, sufferers become increasingly disabled as the disease progresses. If the heart or respiratory system is affected, the condition may be terminal. There are about 60 different forms of muscular dystrophy, ranging from the most common Duchenne MD, which often causes death by the age of thirty, to rarer, more slowly-progressing or later-onset forms.
All types of MD are caused by mutations in genes implicated in muscle structure and function. For instance, Duchenne MD is caused by a mutation in the gene encoding the protein dystrophin, which helps hold muscle cells together, giving them strength. Errors in such genes result in muscle weakness, damage and inflammation, although the severity and speed of disease progression can vary considerably between different mutations.
Dystrophin is actually part of a complex of molecules situated in the membrane encircling muscle cells. Other members of the complex include proteins called sarcoglycans, and mutations in sarcoglycan genes cause another type of MD, limb-girdle MD, which begins in the muscles of the shoulder and pelvis. Dying for a treatment One promising avenue lies in reinforcing the muscle cell membrane in the absence of dystrophin or one of the sarcoglycans. “If we can restore as little as twenty percent of the function of dystrophin, for instance, others have seen real functional benefits for patients,” says Dr Heydemann. One thing all forms of MD have in common is a Her current work focuses on a drug called FTY720 or ‘fingolimod’, already licensed for treatment of chronic inflammation in the debilitating autoimmune disease, multiple sclerosis (MS). Fingolimod – which is marketed under the trade name Gilenya – appears to interact with membrane-bound receptors called the sphingosine-1-phosphate receptors, which are involved in many crucial biological processes, including immune responses. Fingolimod binds to the receptors by mimicking its substrate, sphingosine-1-phosphate. In doing so, it triggers a reduction in the permeability of the cell membrane and strengthens it against damage. Dr Heydemann is testing fingolimod in genetically engineered strains of laboratory mice bred to have and exhibit the symptoms of limb-girdle MD type 2C – a fully lethal disease in humans – and has already found significant effects upon disease progression in three different types of muscle: skeletal muscle, which controls limb movements; heart muscle; and the diaphragm, which facilitates breathing. A multi-functional molecule Dr Heydemann’s mouse studies have shown that just a three-week treatment with fingolimod dramatically inhibits fibrosis in all three muscle types studied. Ultimately, these changes produce measurable and potentially life-changing improvements in the function of the heart and diaphragm. However, treatment must be initiated early: it does not yet appear possible to reverse the changes caused by MD, but only to prevent progression from occurring. Fingolimod is a promising new option for MD treatment: it has few side effects and, being already approved in many countries for MS treatment, it should be possible to move quickly to the stage of clinical trials. Furthermore, the mechanisms by which it fights MD are entirely separate from that of the steroid drugs usually used to treat the disease’s symptoms. This means that the two therapies can be used together in a coordinated approach to produce highly beneficial outcomes for patients. In fact, as Dr Heydemann puts it: “The real advantage of the long list of therapies that are now being developed for MD is that patient-specific co-therapies can be formulated.” These target multiple points in the disease progression and limit side effects by minimising the dosage of any one treatment. There are many reasons to be optimistic regarding the imminently available therapies for muscular dystrophy More questions than answers Intriguingly, initial studies have shown that fingolimod can cause cells to increase production of certain sarcoglycans, suggesting it may be able to re-establish parts of the dystrophin complex. She is also investigating how fingolimod interacts with existing steroid treatments to produce further improvements in patient health. And she is studying a new ‘super-healing’ strain of mouse in which MD symptoms, particularly fibrosis, are diminished. MD research is an exciting field to work in, with so many potential treatments coming to light that it is difficult to attract enough patients to take part in clinical trials for all of them. As Dr Heydemann herself concludes: “There are many good reasons … to be very optimistic regarding the imminently available therapies for muscular dystrophy.” How did you get involved in muscular dystrophy research? What is fibrosis and what is its significance in muscular dystrophy? Why would a drug used to treat multiple sclerosis also be beneficial in the treatment of muscular dystrophy? I feel once these last hurdles are surpassed a large number of young muscular dystrophy patients can begin to have healthier longer lives How important are animal models in your research? What do your results mean for sufferers of muscular dystrophy?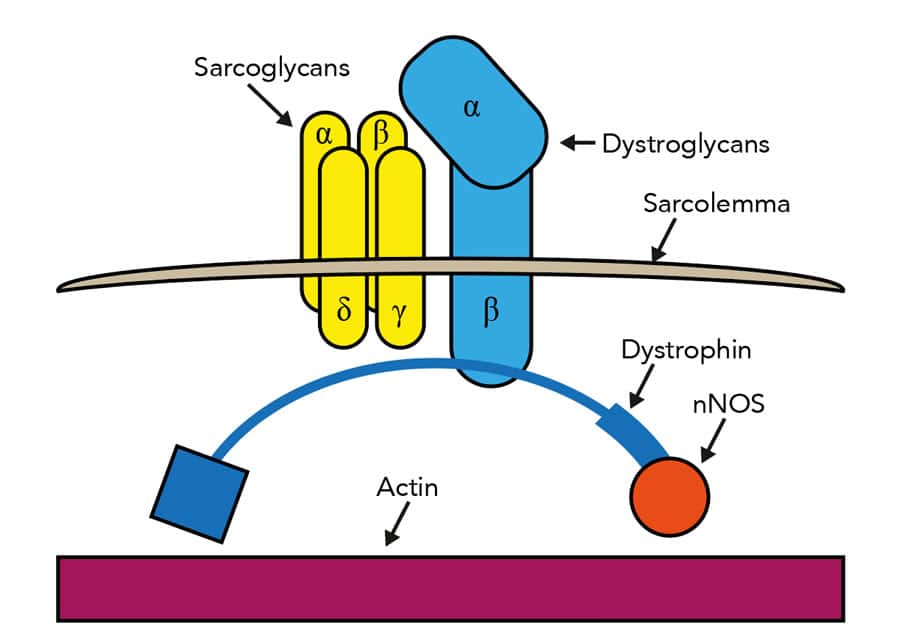
One thing all forms of MD have in common is a lack of effective treatment. Current therapies, often reliant upon steroids, can lessen the symptoms but are unable to slow disease progression. They also come with many unwanted side effects. Dr Heydemann is one of a number of researchers looking at new methods of treating the causes of MD.
lack of effective treatment
Dr Heydemann has documented that fingolimod has at least three separate mechanisms of action against MD. Firstly, it initiates molecular changes which support the membrane surrounding muscle cells. This strengthens the muscles. Secondly, as in MS, fingolimod reduces the chronic inflammation caused by infiltration of the body’s own immune response. Thirdly, it prevents fibrosis, a build-up of inflexible connective tissue which impedes muscle function by preventing the tissue from contracting. Crucially, fibrosis also hinders muscle relaxation, which is vital for the correct functioning of muscles, including the heart and diaphragm. Fibrosis also hinders damaged muscle tissue from regenerating, and impedes the delivery of treatments to affected muscles.
Dr Heydemann’s work into fingolimod continues: so far it has raised almost more questions than it has answered! She now aims to fine-tune the dosage and treatment regime to maximise benefits in her MD mice, as well as investigating any other potential benefits it might have beyond reducing fibrosis and strengthening the muscle membrane. Crucially, she intends to investigate exactly how fingolimod works, including the highly complex cascade of cellular and molecular signals that are brought to play downstream of the sphingosine-1-phosphate receptors.Q&A
This feature article was created with the approval of the research team featured. This is a collaborative production, supported by those featured to aid free of charge, global distribution.
Want to read more articles like this?
Sign up to our mailing list and read about the topics that matter to you the most.
Sign Up!One thought on “New hope for muscular dystrophy treatment”
Leave a Reply
You must be logged in to post a comment.
Hi i have md fsh. Im 46 years old and as far has i could remember i was maybe 12 i new i had it. My mother had,my aunt,myself, one other cousins and last but not least my 23 year daughter. I new the moment i gave her a bottle she had it. Im 46 can no longer walk with out holding the walls also using a walker. My thing is u EVER need to do testing on me but most be all paid for. I will do it in a heartbeat. Research all on me u want! I hate this illness!