Identification of a novel key player in lupus disease opens the door to treatment
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease, the precise cause of which is still unclear. Through dedicated work, Professor Shunichi Shiozawa from Kobe University, Japan, uncovers the underlying cellular and molecular mechanisms of systemic autoimmunity and identifies a novel key player. Shiozawa demonstrates that overstimulation of the host’s immune system by an immunogenic pathogen kickstarts the generation of new T lymphocytes, which induce SLE and could be a future target of SLE cell therapies.
The immune system has two principal lines of defence, namely innate immunity and adaptive immunity. Innate immunity is the body’s first line of defence, providing a rapid and non-specific immune response against microbial aggression. On the other hand, adaptive immunity eliminates specific pathogens or pathogen-infected cells, and provides the basis for protection from infectious diseases. This system has the advantage of immunologic memory, meaning that it ‘remembers’ pathogens the body encountered previously and can respond to them more rapidly and effectively. Coordination between the two systems is essential for the efficient recognition and clearance of pathogens.
Our immune system can be affected by many different types of disorders that impair its function, including autoimmune diseases such as systemic lupus erythematosus (SLE). Unfortunately, we still don’t fully understand the cellular and molecular mechanisms behind this disease. Since the early 1980s, Professor Shunichi Shiozawa and his colleagues at Kobe University in Japan have focused on unlocking the secrets of autoimmunity with extensive experimentation.

Adaptive immune responses
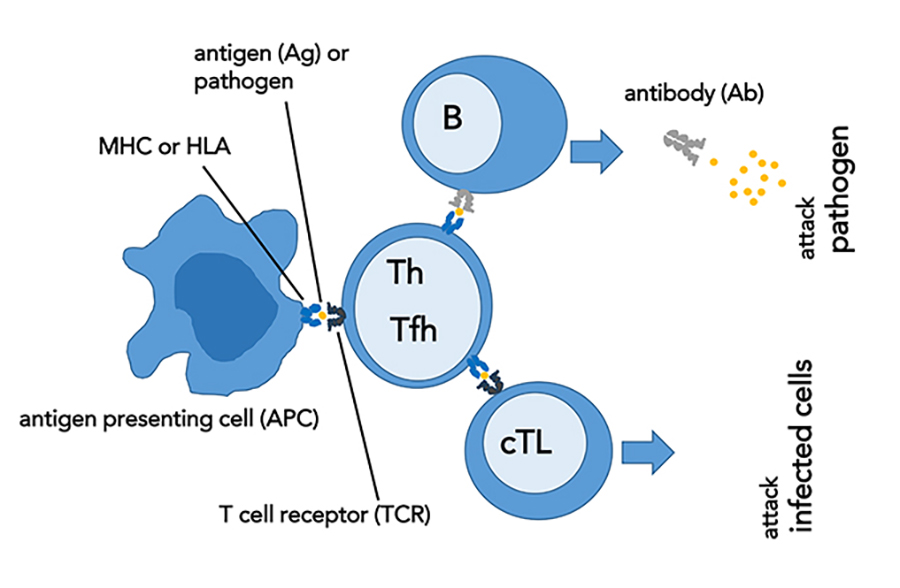
During adaptive immune responses, specific T and B lymphocytes become activated. T lymphocytes derive their name from the thymus, a small gland in the lymphatic system where they differentiate (become specialised). B-lymphocytes differentiate in the bone marrow. All T lymphocytes possess T cell receptors (TCRs), which are randomly generated cell-surface proteins that recognise and bind to foreign antigenic proteins presented by major histocompatibility complex (MHC) molecules. MHC molecules are also called human leukocyte antigens or HLA in humans. TCR recognition of foreign antigen-HLA complex is a prerequisite for the generation of T cell-mediated immunity. TCRs are antigen-specific: upon binding a particular MHC-bound peptide, the T cell possessing the TCR that recognises that specific antigen will proliferate, and its subsequent clones will form. Each individual has an extremely diverse TCR repertoire, which is required for the recognition of a wide range of foreign antigens.
Such a diverse TCR repertoire is created through a random generation process called V (variable), D (diversity), and J (joining) recombination, where gene segments are reassembled by a ‘cut and paste’ mechanism during T cell development. This V(D)J recombination, also called TCR revision, is initiated by recombination activating gene (RAG) complexes, whose expression is restricted to developing T lymphocytes in the thymus. T lymphocytes that have autoreactive TCRs are eliminated, and the TCR revision does not occur after the T cells have left the thymus, because RAG is downregulated by the steady-state mutual interaction between mature T cells. These are the safeguarding mechanisms inhibiting autoimmunity.
Shiozawa and his colleagues have focused on unlocking the secrets of autoimmunity with extensive experimentation.Based on their surface markers, T lymphocytes are primarily subclassified as cytotoxic T lymphocytes (CD8+) and helper T lymphocytes (CD4+). While cytotoxic T cells are critical for the clearance of target cells such as virus-infected cells and cancer-transformed cells, B cells participate in the production of the various circulating antibodies in response to a variety of foreign antigens. On the other hand, helper T cells interact with the aforementioned cells to regulate these cell-mediated and antibody-mediated immune functions. Specifically, a subset of helper T cells, T follicular helper cells, have a profound effect on antibody production by providing the activated B cells mainly in lymph nodes and spleen with growth and survival signals for their subsequent differentiation into long-lived antibody-producing cells.
Dysfunctional T and B cells have been shown to be harmful players in autoimmune diseases, causing the immune system to react against itself. Moreover, although cooperation between T cells and B cells ensures successful immune function, the interaction between these cells is also implicated in the development of autoimmunity.
An overview of lupus disease
SLE is a multisystemic autoimmune disease, affecting multiple organs and tissues such as the brain, blood, and kidney in most patients. Improvements in treatment with a smarter use of immunosuppressive drugs and more efficient treatment of infections have significantly increased the 10-year survival rate to more than 70% over the last 50 years. Nevertheless, current management strategies show limited benefits. Corticosteroid therapy, for example, is frequently associated with long-term adverse effects.
Originally, lupus was considered a skin disorder. The word ‘lupus’ is derived from Latin for ‘wolf’ and was used to define the skin lesions, which resembled a wolf’s bite. Later, it was realised that lupus erythematosus is a systemic disease, characterised by defects in immune responses causing inflammation and damage in various organs. In 1949, researchers Mackay and Burnet proposed their autoimmune disease theory, which suggested that immunologically competent cells or antibodies attack the normal components of the body. Although this initiated extensive research since, the precise mechanisms of SLE were still not completely understood. Substantial research focused on the abnormalities of pathogens, including autoantigens, as the cause of systemic autoimmunity, yet the pathogenesis (the mechanism of the development) of SLE remained a puzzle.
What is the cause of systemic autoimmunity?
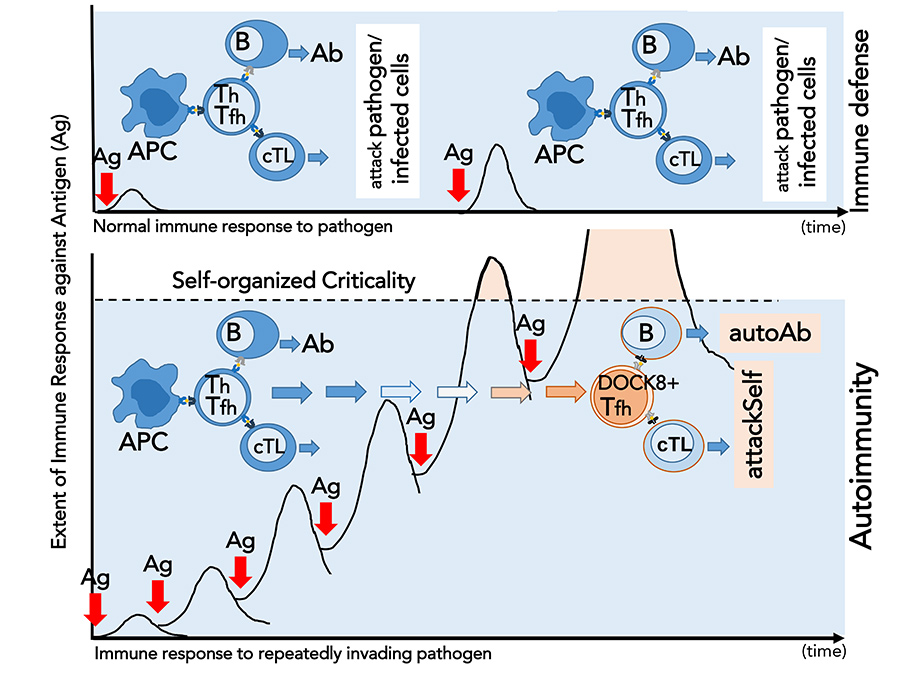
In 2009, Shiozawa and colleagues described a novel approach to researching the pathogenesis of autoimmunity. Unlike previous studies, they investigated the integrity of the immune system. The researchers overstimulated the immune system in mice that are not prone to autoimmune diseases by exposing them repeatedly to an antigen and successfully induced the development of SLE. In other words, the overstimulation of the immune system by external disturbance disintegrated the steady-state immune response by surpassing its stability limit, ultimately leading to systemic autoimmunity.
Based on their results, Shiozawa and colleagues proposed the ‘self-organised criticality theory’ to explain the pathogenesis of SLE. According to this theory, autoimmunity does not occur when the immune system is stable and its integrity is maintained. However, when repeated immunisation with an antigen surpasses the stability limit of the immune system, systemic autoimmunity takes place. Another important finding is that no specific antigen is required for autoimmunity to take place.
Shiozawa’s years of dedicated work offers new insight into the mechanisms of systemic autoimmunity.Furthermore, Shiozawa and colleagues identified novel T lymphocyte clones that are responsible for the induction of SLE. They demonstrated that repeated exposure to an antigen induces the development of autoreactive T lymphocyte clones through the revision of their TCRs. These novel T cells, which Shiozawa named ‘autoantibody-inducing CD4 T cell’ (aiCD4 T cell), have been shown to be capable of stimulating both cytotoxic T cells and B cells. Thus, aiCD4 T cells direct B cells to secrete autoantibodies including anti-DNA antibodies, which are a characteristic of lupus. In addition, aiCD4 T cells stimulate the maturation of cytotoxic T cells into effector (‘fighter’) cells that are required for their cytotoxic actions and tissue injury.
SLE is characterised by various cellular and molecular abnormalities, including the formation and deposition of autoantibodies and antigen-antibody complexes, named immune complexes (ICs) in various organs. In 2013, following the identification of the aiCD4 T cell in the pathogenesis of SLE, Shiozawa and colleagues further investigated the mechanisms of autoimmune tissue injury in the kidney with a specific focus on the effector T lymphocytes. The results showed that autoimmune tissue injury is caused by the cytotoxic T cells, which matured into effector cells with the help of the aiCD4 T cells. These cytotoxic T cells recognised antigen presented as IC on the target tissue and induced renal disease.

Characterisation of the novel T lymphocyte
In their latest work, Shiozawa and colleagues paid specific attention to the aiCD4 T cell, which is directly linked to the synthesis of diverse autoantibodies by activated B cells and the maturation of effector cells during autoimmune tissue injury. They isolated and characterised this key player to uncover the mysteries of SLE pathogenesis, as well as to propose a potential therapeutic target. The molecular and biochemical analysis demonstrated that aiCD4 T cells are T follicular helper (Tfh) cells, and uniquely express the guanine nucleotide exchange factor DOCK8 on the cell surface, now called DOCK8+Tfh cells. The Tfh cell is a principal conductor of the immune response, directing antibody formation by B cells and regulating cellular cytotoxic activity. The transfer of these DOCK8+Tfh cells to healthy recipient mice resulted in the generation of a variety of autoantibodies and organ diseases such as renal (kidney) disease; it also led to the generation of lesions in organs other than the kidney, primarily skin, lung, and spleen, showing that these cells initiate SLE.
The key finding is that the TCR of DOCK8+Tfh cells are newly generated; this results in novel, autoreactive T lymphocyte clones, which induce SLE. Shiozawa showed that when the steady-state response is disrupted, that is to say, the T cell response surpasses the immune system’s self-organised criticality, RAG gene complex initiating the V(D)J recombination is reexpressed. In DOCK8+T follicular helper cells, TCR gene chromatin becomes accessible to transcription factors such as E2-2 or DOCK8, and novel TCRs are generated. Thus, DOCK8+Tfh cell is a novel T follicular helper cell, which underwent TCR revision in the splenic red pulp and became an autoreactive T cell with the capability to initiate the production of a variety of autoantibodies and the development of SLE. They also found that these novel DOCK8+Tfh cells are significantly increased in the spleen and peripheral blood of patients with active SLE.
Pathogens that induce SLE
Living organisms are continuously exposed to various pathogens, including non-fatal viruses, which can infect the host if they evade the cytotoxic T cell responses. An example of continuous exposure to pathogens might be the measles virus infection, associated with the recent outbreak in Japan in a subpopulation of young adults who avoided vaccination against the virus. The virus may not induce an apparent immune response due to the low levels of cytotoxic T cells in memory at the time of the reinfection. On the other hand, repeated exposure to such an antigen in combination with appropriate host HLA may overdrive the immune response to surpass the immune system’s self-organised criticality, thereby leading to the development of the novel DOCK8+T follicular helper cells, which then induced SLE by the production of diverse autoantibodies and the cell-mediated response.
A potential therapy for SLE
Most recently, Shiozawa and colleagues have presented a potential SLE therapy in their latest research. They demonstrated decreased production of autoantibodies, improved kidney histology, and reduced lesions of the skin, lungs, and thyroid following the treatment with DOCK8 antibody targeting the DOCK8+T follicular helper cells, the key players in SLE pathogenesis. Thus, Shiozawa and colleagues’ years of dedicated work offers new insight into the mechanisms of systemic autoimmunity with the presentation of the ‘self-organised criticality theory’. Identification and characterisation of the DOCK8+ T follicular helper cells not only shed light on the SLE pathogenesis, but also revealed a possible target of SLE cell therapies.
Personal Response
What is the potential of the DOCK8 antibody to become a therapeutic agent to treat SLE?A host’s immune response is a system response, and every system response has its steady-state and its limit, called self-organised criticality. As to the cause of autoimmunity or SLE, not a particular pathogen is responsible. Instead, when an immunogenic pathogen presented on HLA repeatedly stimulates the host’s immune system to levels beyond self-organised criticality, system response is disrupted. As a result, T cells recover from the state of ‘unrespons iveness’ and new autoreactive T lymphocyte clones are generated via de novo TCR revision from the non-autoreactive cells, which have passed the selection process in the thymus during their development. These newly emerging cells are the novel DOCK8-expressing T follicular helper cells, which now stimulate B cells to produce varieties of autoantibody and mature cytotoxic T cells to induce lupus disease. Anti-DOCK8 Ab treatment ameliorated SLE in mice