Drug-adapted RET mutations
Protein tyrosine kinases (PTKs) are enzymes that cause protein tyrosine phosphorylation, a process that regulates proteins and leads to further downstream effects as they carry out their function. PTKs are key players in many cellular interactions and reactions, known as signalling pathways, which cause changes at the gene level. Disruption to them can therefore lead to major health problems.
Sometimes specific genes, known as oncogenes, become aberrantly activated due to these disruptions. This can cause tumours as upregulated activation of an oncogene can promote cell proliferation and survival, both of which are hallmarks of cancer. This is the case when the protein tyrosine kinase RET (REarranged during Transfection) is activated in an uncontrolled way.
Aberrant RET activation
RET is a transmembrane PTK which is important in foetal development of various areas of the body, such as the kidneys and the enteric nervous system. RET is normally activated by binding specific ligands, which induce a homodimerisation of the transmembrane protein and its co-receptor complex, where a protein complex is formed from two identical proteins or protein complexes. This allows the two RET PTK to undergo autophosphorylation and subsequent activation of the signalling pathways. RET can activate several signalling pathways, particularly the RAS/MAPK pathway, which is affected in many cancers.
Acquired resistance to these new drugs seems to be an inevitability based on repeated experiences with previous therapies.
In normal circumstances, RET activation is regulated, but genetic changes can occur that lead to the protein becoming constantly active. RET can become altered either as a result of gene fusion or by point mutations, some of which are hereditary in families with predispositions to multiple endocrine neoplasia 2 (MEN2) – a condition affecting endocrine glands. Both of these changes lead to uncontrolled RET activation which, in turn, means that further reactions in the signalling pathway will also be switched on. Downstream effects of this are the increased proliferation and survival of cells, which causes tumours to form.
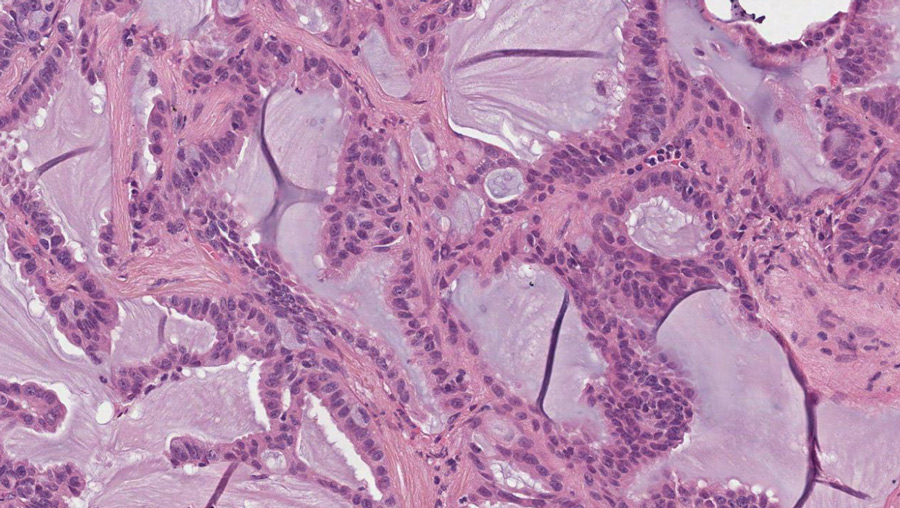
This aberrantly active RET protein has been seen in many types of cancer, including medullary thyroid carcinoma (MTC), papillary thyroid cancer (PTC), and non-small-cell lung cancer (NSCLC), amongst others. Laboratory experiments have verified that the genetically changed RET gene is the cause of these cancers and that abnormal RET kinase activity is required to keep the tumour cell alive. This makes RET a good target for cancer treatment.
Targeting PTKs
Due to the oncogenic nature of many PTKs, they have been used as patient-specific targets in treating cancer. Many PTK inhibitors exist, such as EGFR inhibitors, and are used in clinical settings. They work by directly binding to the active site of the enzymes so that they can no longer phosphorylate proteins, thereby inactivating them, leading to anti-tumorigenic effects. Before RET-specific inhibitors could be established, multi-targeted tyrosine kinase inhibitors were developed for other kinases, such as vandetanib and lenvatinib. These inhibitors were used against RET-altered cancers with some success; however, they can lead to off-target effects, and resistance has been seen in some cases.
Structural studies have revealed that these types of PTK inhibitors bind to the front and back of the drug-binding clefts of RET by going through the gate region between these two clefts. However, some cancer cells can adapt to these inhibitors by substituting an amino acid called the gatekeeper with a different amino acid. This substitution has been shown to confer resistance by interrupting the binding of the inhibitor to the RET molecule, allowing it to become activated again and the cancer to progress.
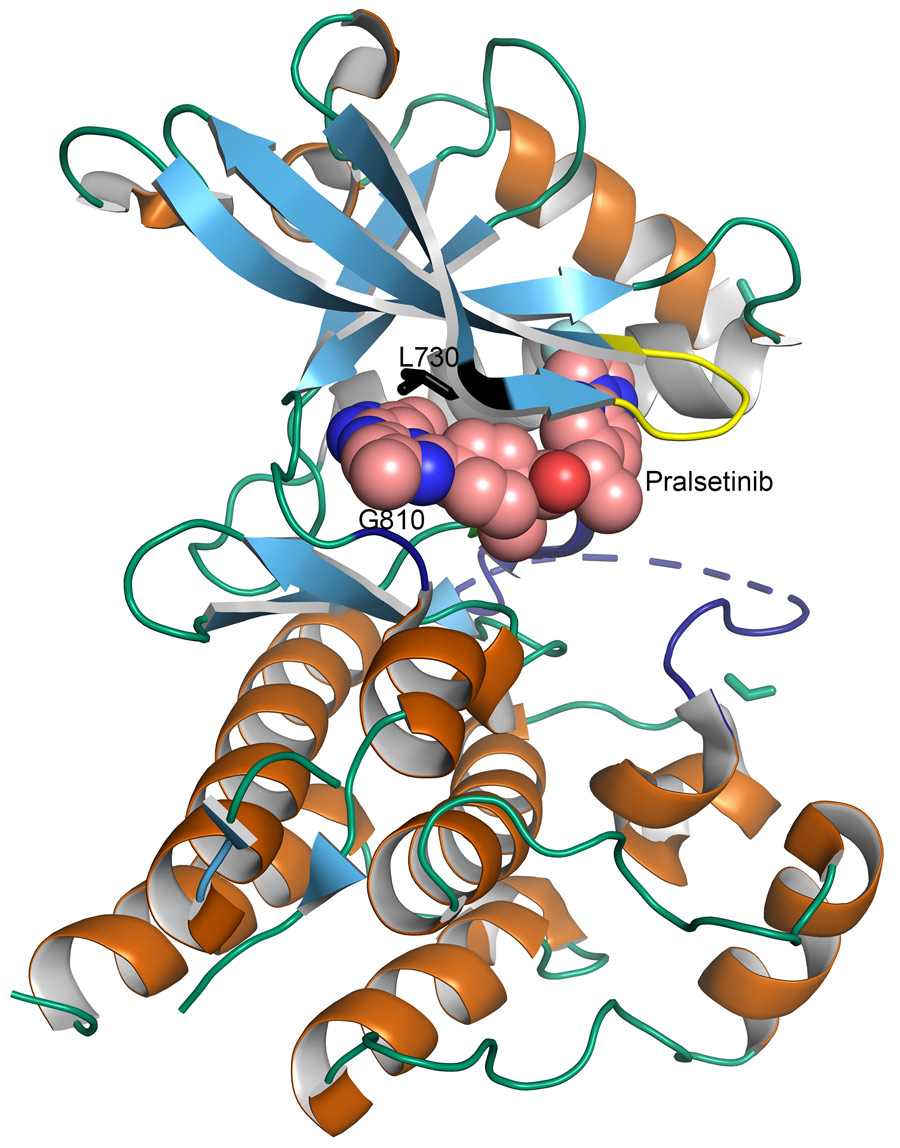
Pralsetinib and selpercatinib
Pralsetinib and selpercatinib are new PTKs designed specifically to inhibit RET. Compared with the older, multi-targeted PTK inhibitors, pralsetinib and selpercatinib are much more potent and selectively inhibit the RET kinase activity. This results in more effective inhibition of RET kinase activity in the tumours, and fewer side effects. The US FDA recently approved pralsetinib and selpercatinib for use against RET-altered NSCLC, MTC, and PTC, and they have also been approved in Europe. Most importantly, they have been used effectively for patients who had developed resistance to the more general PTK inhibitors. This is because pralsetinib and selpercatinib bind to RET in a very different way that avoids interference by changes in the gatekeeper amino acids.
Crystal structures were determined by Dr Mooers and colleagues at the University of Oklahoma Health Sciences Centre from high-resolution diffraction data collected with synchrotron radiation. These structures have shown that pralsetinib and selpercatinib bind both the front and the back pockets of the active site, which is similar to the other inhibitors. The difference is that they do not go through the gate region and instead wrap around it. This means that the gatekeeper mutations seen in resistant cancers would have no effect on pralsetinib and selpercatinib. These inhibitors are still able to bind and inactivate RET in the presence of gatekeeper mutations. However, these inhibitors interact with several non-essential amino acid residues of the RET kinase. This makes pralsetinib and selpercatinib vulnerable to drug-resistant mutations in these amino acids.
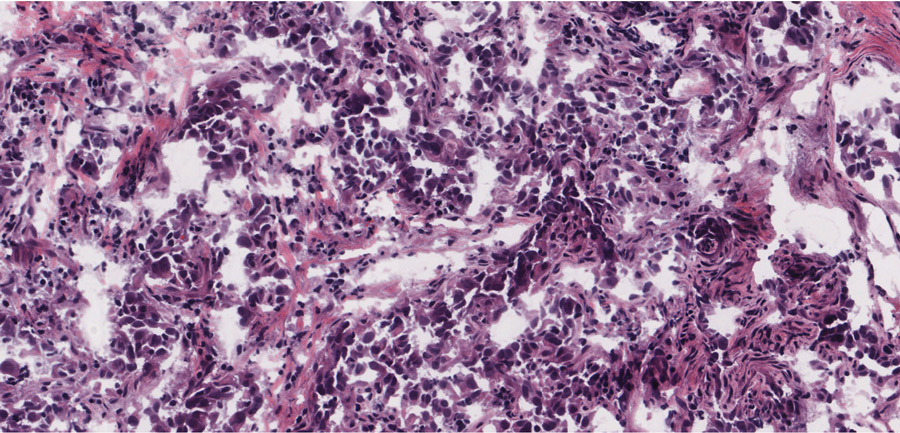
Further resistance being seen
The new RET-specific drugs seem to be working much more effectively than the more general inhibitors, with response rates of 60–85% over 18 months. However, acquired resistance to these new drugs seems to be a certainty based on repeated experiences with previous therapies. We need to be always mindful of acquired resistance. For this reason, Dr Wu and his colleagues decided to take a proactive approach by determining where resistance could arise from.
Understanding how resistance is conferred is the key for the future of RET-specific treatment.
By carrying out experiments on various cell lines, Dr Wu was able to discover mutations that conferred resistance to both selpercatinib and pralsetinib. These cell cultures were treated with increasing concentrations of the drugs which eventually led to the acquired resistance. Dr Subbiah has been treating and following up patients with RET-positive cancers enrolled on these novel RET-inhibitor clinical trials. Most patients respond to the therapy, indicated by tumour shrinkage in the scans and improved quality of life. However, after around two years, the drugs stop working. This is because cancer develops mechanisms that render the drugs ineffective. These are known as acquired resistance. The mechanisms of resistance seen in the structures by Dr Mooers and Dr Wu were also seen in the tumours that became resistant to therapy. In some cases, RET had been bypassed altogether by activating a downstream RET target. In other cases, there were direct drug-adapted mutations. As mentioned before, these inhibitors do not bind the gate region of RET, so they were able to overcome resistance by the gatekeeper mutations. It would seem, therefore, that non-gatekeeper mutations were responsible for the resistance seen in patients and cell cultures.
The resistant mutants from the cell cultures were sequenced, as was cell-free DNA from the patients who had acquired resistance. The mutations were determined to be in the solvent front, the hinge, and the 2 strand areas of RET. All of these areas are involved in the binding between selpercatinib or pralsetinib with RET. The strongest resistance to both drugs was seen to be conferred by substitutions of the glycine 810 residue in the solvent front region, which implies that the RET with glycine 810 mutations will be the least inhibited by these drugs and particularly important for causing acquired resistance.
The future of RET therapy
Drug resistance is always a problem when developing a new target in cancer therapy. It is therefore important to take a proactive stance and predict what mutations could occur before they even develop in patients. This way, the next generation of drugs may be developed earlier to circumvent the resistance. RET is clearly an extremely good target as it is mutated in so many cancers, and inhibiting it has such promising results in patients. Being able to predict what could happen or understanding why resistance is occurring is vital to keeping treatments as effective as possible. More RET-specific drugs are in the works, and the possibility of using these in combination with others is promising. Understanding exactly how the drug works and how resistance is conferred is the key for the future of RET-specific treatment in order to keep one step ahead of cancer.
Personal Response
Could combination therapy with RET inhibitors be used to overcome this resistance?
In metastatic cancers, residual tumours that cannot be completely eliminated by a cancer treatment will evolve to become resistant tumours, leading to disease progression. Identification of drug-adapted mutations in protein tyrosine kinase-targeted cancer therapies is essential for developing the next generation of drugs to inhibit these mutants, in order to turn an incurable malignancy into a manageable chronic disease.