NMR2: A highly accurate approach to protein-ligand binding
Proteins are the fundamental building blocks of all living matter, from microscopic viruses and bacteria to highly evolved multicellular organisms, plants, animals and humans. They play a role virtually in all phenomena associated with life, including providing structural support to individual cells and tissues, enabling motion in complex organisms, producing energy and regulating signalling between cells in the body. Frequently, the physiological function of proteins is modulated by the interaction with small molecules (ligands), which can bind to specific receptor sites in a protein and trigger a response, e.g. in the form of a structural change or of a chemical reaction. For instance, ligands like hormones can promote cell growth by increasing the rate of protein production or they can induce relaxation in muscle tissue. Similarly, cofactors are ligands that play an essential role in a number of complex chemical reactions involved in the metabolic processes that keep an organism alive. Most drugs can themselves be classified as artificial ligands. They work in the same way as natural ligands, by binding to specific sites in proteins and modifying the protein’s function.
A famous example of this is penicillin (an antibiotic, and one of the first medications found to be effective against many bacterial infections caused by staphylococci and streptococci), discovered by Alexander Fleming in 1928. Penicillin acts by binding to receptor sites in bacterial cell membrane proteins, which drastically affects the bacterial cell wall growth and eventually leads to its degradation and to bacterial death. Some anti-cancer drugs act in a similar way, by blocking the proteins responsible for DNA synthesis and hindering the growth of new cancerous tissue. Understanding how proteins involved in bacterial infections or ontogenesis are affected by specific drugs, and how we can optimise these drugs to maximise their effects is currently one of the crucial and most far-reaching issues in pharmacological research and drug discovery. Protein structure determination Understanding the nature of the interactions between proteins and ligands is a crucial issue in drug discovery. This approach is very error-prone and it requires validation, e.g. by comparison to refined 3D complex structures. The gold standard in 3D protein structure determination is currently provided by X-ray crystallography, a powerful approach that is frequently used to work out the structure of protein-ligand complexes, essentially at an atomic level. This technique requires crystalline samples, which can be expensive and time-consuming to obtain. X-ray crystallography can also run into problems with specific classes of proteins, including membrane proteins and flexible receptors. The efficiency of X-ray crystallography can be improved substantially using the Molecular Replacement (MR) method, which relies on the existence of a previously resolved structure that is similar to the protein-complex under study. However, some classes of proteins (like membrane proteins and flexible receptors) remain beyond the capabilities of X-rays, and a radically different technique, nuclear magnetic resonance (NMR), has been proposed as a potential alternative for these cases. NMR uses magnetic fields to gain information about the environment of each individual atom in a protein, for example, its chemical shift and dipole-dipole interaction. The main limitation of NMR is that this technique requires long measurements and extensive data analysis. A recently proposed and fast-developing technique, cryo-electron microscopic, has also been shown to have great potential in drug discovery for large systems. Currently, however, X-ray crystallography and, to a lesser extent, NMR are by far the most widely used approaches to structural determination, and hundreds of thousands of protein structures have been resolved to date using these two complementary techniques. The new method developed by Dr Orts and his collaborators aim to bridge the gap between X-rays and NMR, in order to expand the power and applicability of NMR and to make it a robust tool for drug discovery. NMR2: a zoom on the binding sites A typical NMR2 protein-ligand structure determination requires a preliminary preparation of the sample, in which either the protein or the ligand are isotopically substituted (13C, 15N) or selectively labelled (e.g. isoleucine, leucine and valine methyl labelling). NMR experiments are then used to measure intra-molecular (ligand) and inter-molecular (ligand-protein) atomic distances, which in turn provide a model of the ligand structure in the binding pocket. To understand the exact nature of the ligand-protein interaction, protein structures from existing databases (obtained from X-ray or NMR measurements) are then used as input information. The structures selected can be either that of the protein in the absence of the ligand or those of similar (homolog) proteins. The NMR2 methods reduce the time required to determine protein-ligand structures from months to a few days. The NMR2 program then screens all possible assignment groups in the protein and calculates the protein-ligand complex structure for all options. At this stage, it is essential to reduce as much as possible the number of configurations to screen. This can be achieved by initially restricting the assignment groups in the protein to only 3 or 4 relevant ones. False assignments can be ruled out using geometric considerations, based on the knowledge of the input structures. This substantially reduces the calculation time. At the end of this procedure, the resulting complex structures have to be analysed carefully, to detect potential errors arising from the unconstrained relaxation of the protein backbone during the refinement procedure. It is important that a sufficient number of inter-molecular distances are taken into account, typically at least 12 or 15. A high signal-to-noise in the NMR spectra and a good signal resolution are also crucial. A new tool for drug discovery The accuracy and efficiency of the NMR2 methods have been documented in a number of protein-ligand complexes. What are the remaining challenges of NMR2 that need to be addressed in order to make your method a robust and easy to use routine tool for high-throughput molecular screening in drug discovery? Having access to the structure of the binding site for each binder allows investigating chemical scaffolds that would otherwise be discarded and to broaden the chemical knowledge as well as the drug-ability of the receptors.
Proteins are typically large and complex macromolecules, composed of thousands of atoms, which are arranged in chains of subunits, called amino acids. Proteins fold into 3D structures, which are characteristic of each protein and are connected at a fundamental level to the protein’s function. Accurate knowledge of a protein’s structure is the first step toward understanding how the protein works and how the chemico-physical processes that it carries out can be influenced by means of drugs. Nowadays, drug discovery typically starts by screening large databases of molecules or molecular fragments as potential ligands for a selected drug target.
The protein-ligand binding arises from very local interactions: ligands only bind to very specific sites in a protein, which typically involve only a relatively small number of atoms compared to the whole protein. Binding sites are just like pockets in the protein, which a ligand can enter and in which it can be stabilised by steric (i.e. depending on the shape and size of the pocket), electrostatic or chemical interactions. The global structure of the protein is initially largely unaffected by the ligand binding. Based on this observation, Dr Orts and his collaborators have developed a powerful protocol to work out in great detail the structure of the protein-ligand binding site, using existing information about the global structure of the protein obtained in separate X-ray diffraction or liquid NMR measurements. This method, the Nuclear Magnetic Resonance Molecular replacement (NMR2), is based on solution-state vMR structure determination, but it focuses on the structure of the binding-site alone, rather than on the full protein, and therefore it circumvents the lengthy and tedious analysis required to resolve the whole protein structure from NMR data. NMR2 drastically reduces the time and effort required to obtain an atomically resolved structure of a binding pocket by using previously determined protein structures, from a couple of months to a couple of days. It can also be partially automated, and therefore it provides a natural tool for high-throughput workflows, which can be used to screen thousands of potential new drugs in sequence and to analyse the nature of their interaction with a target protein.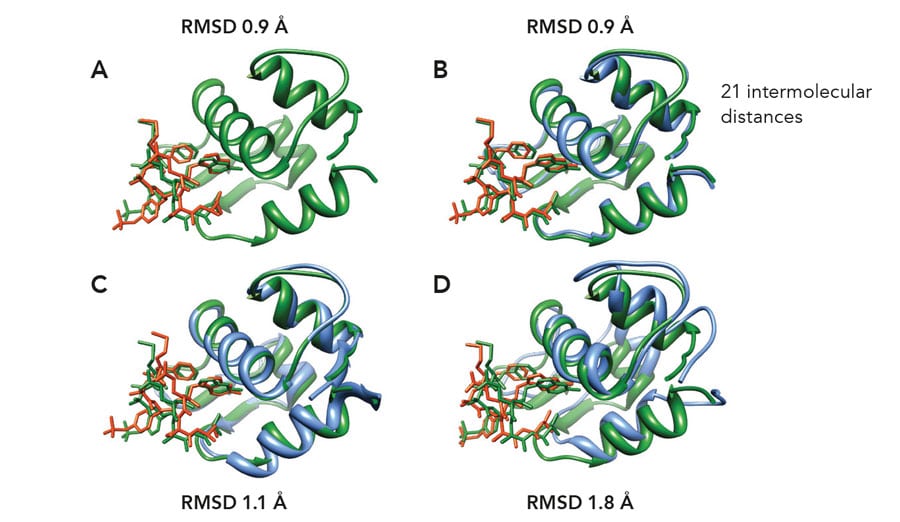

The NMR2 method has been applied successfully to the resolution of various classes of protein-ligand complexes. Several structures containing strong binders or small ligands have been determined with an accuracy of 0.9-1.5 angstroms relative to the reference structure. The applicability of NMR2 to complexes with ligands in fast exchange or weak affinity binding has also been demonstrated. In the case of the weak affinity binding complex HDM2-#845, a new complex has been characterised, never observed before. The efficiency NMR2 is well represented by the complex structure of SJ212-MDMX, which could be resolved at 1.35-angstrom accuracy within a day using a desktop computer. These are a few initial examples of the great potential of NMR2 in the study of protein-ligand interactions and protein function and they pave the way for its application as a fast, reliable and accurate protocol for drug discovery.Personal Response