Quantum molecular modelling for efficient chemical synthesis
The modern world is full of things designed on computers: your car, your toaster, your house and your computer itself. Computer Aided Design (CAD) is an essential tool for creating, modifying and optimising designs in civil and electronic engineering. But what about the chemical engineers creating pharmaceuticals, cosmetics and other synthetic chemicals – can they utilise CAD? Increasingly, the answer to that question is “yes”.
The big difference between designing buildings and chemical reaction mechanisms is the quantum mechanics involved in modelling molecules: the bricks and mortar of the chemical world are atoms with slightly blurry edges, and electrons with probability densities rather than definite locations.
Microkinetic modelling
Modelling chemicals and their interactions – known as microkinetic modelling – is tricky, but there are in fact many methods. One of the most successful, Density Functional Theory (DFT), is a utilisation of quantum mechanics to investigate the movement of electrons around multiple atoms. This technology is still developing, and recent progression in DFT software allows synthetic chemists to make complex predictions of how molecules interact. These analyses give a deeper level of insight into chemical processes than ever before, allowing chemists to choose effective catalysts and predict the effect of additives on a reaction.
Recent progression in the combined use of DFT and microkinetic software allows synthetic chemists to predict the outcome of complex reaction systems.
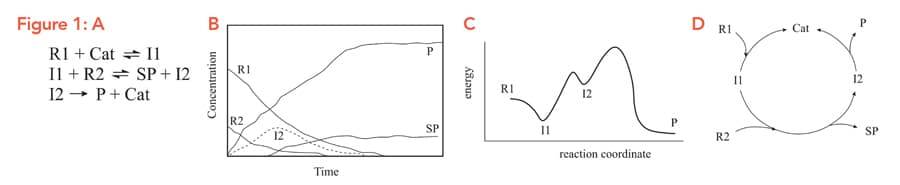
Synthetic chemists are interested in producing new molecules and improving current production methods. These reactions, Figure 1A, often include multiple starting materials and products, and a successful computer analysis of a reaction will need to consider all of these, alongside solvents, catalysts, pre-catalysts and side-products. Over the course of a reaction, Figure 1B, these molecules will go through various ‘transition states’ (TS), Figure 1C, all of which also need to be accounted for. If that’s not enough, catalysts – substances which don’t get used up in the reaction – are regenerated, creating cycles, Figure 1D. Taking all of these into account forms a large network or ‘reaction scheme’ of chemicals and transition states, linked by reversible or irreversible reactions.
Typically, researchers using DFT methods start with a suspected full reaction scheme, which may contain more than 100 individual reactions. The energetics of each reaction within the scheme determine how easy it is for that reaction step to proceed. In the standard protocol, the majority of energetic parameters are fixed at the DFT calculated value, leaving a limited number of parameters that can be varied within the error boundaries of the calculation until the simulation matches experimental values. This process can be time-consuming, due to the computational load of calculating a large number of DFT parameters.
Tightening up the procedure
Over the last three years, Professor Martín Jaraíz at the University of Valladolid in Spain has been using another method – one which provides not only equally or even more accurate and reliable results, but also requires less experimental input and computation time. Rather than starting with every possible reaction pathway, instead begin with just the dominant mechanisms – Jaraíz calls these ‘loose’ and ‘tight’ modelling approaches. Starting with less information may seem counter-intuitive, but because fewer reactions are being modelled, all the parameters can be adjusted in the DFT process. Once the major mechanisms are in place, important additional reactions can be added to fine-tune the model based on experimental data.
Building a model made up of many individual chemical reactions requires specific methods and tools of its own. In the laboratory, Reaction Progress Kinetic Analysis (RPKA) can be used to identify rate laws of the dominant mechanisms – that is, how quickly chemicals are converted into other chemicals. Outside the lab, theoretical approaches like Automated Reaction Path Searches can elucidate the important pathways and transition states. In combination, these can be used to build a reaction scheme of the various reactions that eventually lead to the synthesis of a molecule. The tight method requires initially identifying a minimum number of reactions that can explain the overall reaction.
Once a basic reaction scheme has been proposed, the next step of the DFT process is to consider more deeply the energetics of each reaction step. If all the reactions in a mechanism are energetically plausible, then the proposed mechanism gets the ‘all clear’. On the other hand, models containing reactions which are energetically unfavourable – reactions which are too tricky – are unlikely to represent reality. It is also important to consider the effect of fluctuating levels of chemicals on reversible reactions and their transition states.
Once each reaction in the initial model has earned its place in the scheme, additional information can be incrementally added to the model for fine-tuning. These details are valuable for explaining experimental spectroscopic observations, such as the slow build-up of particular molecules over time leading to catalyst deactivation.
Verifying a proposed model
Professor Jaraíz and his team applied this new protocol to a series of case studies. The first of these is a reaction involving titanocene monochloride (Cp2TiCl), a catalyst sometimes used in ring-opening reactions of epoxides. A mild acid called collidine hydrochloride (CollHCl) is often added to this reaction, which prevents the Cp2TiCl from being deactivated over the course of the reaction. The resulting reaction is highly efficient and converts almost all of the substrate into the desired product, with only a small amount of unwanted side-product. Catalyst deactivation is a very important factor when deciding upon catalytic processes for large-scale industrial chemical synthesis.

Jaraíz and his team began their DFT modelling process with a reaction scheme (Fig. 2A) of five key reactions making up the catalytic cycle. The initial scheme did not take into account catalyst deactivation and the crucial role of CollHCl to suppress deactivation, in order to begin with a simpler reaction system for DFT. Only once the energetics of these reactions were established (i.e. calculated and fine-tuned), Jaraíz modified the reaction scheme by adding steps to account for deactivation (Fig. 2B) and the effect of the CollHCl additive (Fig. 2C).
The use of predictive, DFT-based reaction simulators could have a significant economic impact on the chemical industry.
To test the predictive capability of the procedure, the model was trained on only three experimental data points (Fig. 2A), and the data was collected from a reaction performed without CollHCl. The model was used to predict the results from the synthesis done with and without CollHCl. The model was very successful: it could very accurately predict the shape and yields for this (Fig. 2 A,B) and several other reagents.
For another case study, even the as-calculated DFT parameters predicted correct yields, the only difference with the simulation was that it predicted the reaction to occur about 360 times faster than it actually does. After fine-tuning the DFT parameters, the simulator could match the timescale for the reaction as well.
Proposing a reliable modelling protocol
Another case study illustrates the potency of the ‘tight’ method and how it can provide more reliable results (Fig. 3). For this case study, Jaraíz chose as a representative example a reaction from a recently published reaction scheme, proposed by another research group: the ring opening reaction of 1,2 epoxyoctane by 2 propanol, catalysed by fluorinated aryl borane (FAB).
This reaction is interesting because the presence of unwanted water can cause the regioselectivity of FAB to decrease, resulting in lower amounts of the desired product. A reaction scheme had been previously proposed by another research group, created using the often used ‘loose’ modelling approach. To explain the effect of water on the reaction, the scheme contained 130 reactions, resulting in 149 DFT parameters. Given the large number of parameters, only nine were adjusted, and the remainder were set at the calculated DFT values.
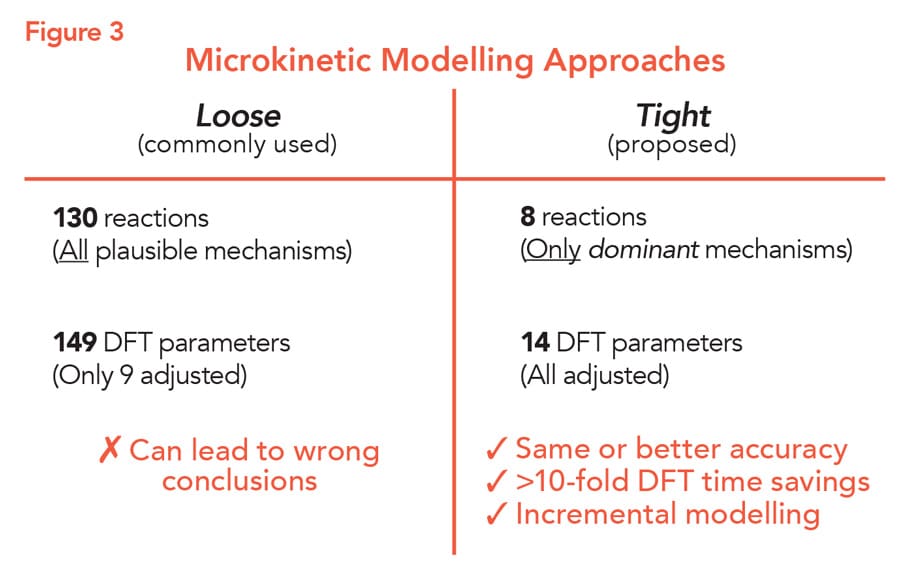
For a tight modelling approach, Jaraíz chose eight for the initial scheme of 130. This resulted in a reduction in the number of DFT parameters to 14, all of which could be adjusted. This initial reaction scheme included the catalytic cycle of FAB to convert the substrate into the product, two deactivation reactions involving FAB, and two reactions creating side-products. The simulation was fine-tuned using only data from five experiments. By this point, the simulation could predict the relationship between the amount of water and both the initial rate and regioselectivity of the reaction with high accuracy.
The biggest discrepancy between the tight and loose models is in the relationship between water level and regioselectivity. The loose model predicted a much stronger relationship than is actually seen in experiments, whereas the tight model provides more accurate values and did not include some unconventional water mediated reactions of the loose model. The tight model deems these steps unnecessary and, thus, reduces the chances of, for example, predicting multiple catalytically relevant species and multiple catalytic pathways whose existence is questionable and that result from the large number of reactions that have only been loosely adjusted and tested.

Chemical CAD
In summary, the tight protocol proposed by Prof Jaraíz is a highly effective substitute for the previous protocol that can provide more robust and reliable models and also drastically decreases computation time. CAD can increasingly be used for chemical synthesis, and this novel implementation of DFT-based kinetic modelling is another step towards fully predictive CAD for chemical processes.
Personal Response
To what extent do industry use predictive techniques like DFT, and in what ways do they stand to benefit from more implementation of these methods?