Cilia, asymmetry, and genetic disease
Left–right asymmetry is a phenomenon widely observed in nature. While many animals may appear symmetrical, asymmetry is pervasive in every major subset of the animal kingdom and can be as subtle as the coiling direction of a snail’s shell, or as evident as the size difference of a fiddler crab’s claws. Chirality – the asymmetrical property of an object which is non-superimposable on its mirror image – is intrinsic in biomolecules such as amino acids and sugars, as well as the majority of cells. However, the mechanisms that produce macroscopic asymmetry in an organism’s tissues or entire body, starting from a symmetrical bundle of embryonic cells, are complex and not fully understood.
The left–right organiser and Pkd2
The left–right organiser is a unique embryonic organ that forms during early development in vertebrates. It establishes left–right organisation within the body, for example the placement of the internal asymmetric organs, such as the liver and the heart. This is achieved by the movement of microscopic hair-like organelles known as cilia, which rotate clockwise to propel embryonic fluid from right to left through the ventral node (a liquid-filled cavity near the centre of the developing embryo). The remarkable consequence of this directional fluid movement is the breaking of symmetry within the embryo; altered calcium ion transients in the cilia and cells around the left–right organiser influence asymmetric gene expression, ultimately causing asymmetric development of the internal organs.
Polycystin 2 (Pkd2), is a protein (TRPP2, polycystin-2, or PKD2) of the transient receptor potential (TRP) superfamily of ion channels. It is involved in cell signalling and has been linked to the perception of flow dynamics. Pkd2 is believed to form part of the cell and ciliary structure that senses the fluid flow in the left–right organiser. In the mouse model, this is facilitated by the existence of a second, non-motile type of cilia, known as mechanosensory cilia, which act as ‘antennas’, detecting changes in the flow outside the cell. Mechanosensory cilia and Pkd2 in mice are vital components for asymmetric development, and an imbalance or absence in either has been shown to lead to left–right defects in mice which can sometimes be fatal.
The mechanisms that produce macroscopic asymmetry in an organism are complex and not fully understood.
The Lyoscil Project
The aim of the LYSOCIL Project, led by researchers at NOVA Medical School, Lisbon, is to gain an understanding of the underlying causes of diseases affecting the cilia, known as ciliopathies. Many of these diseases are very rare, for example primary ciliary dyskinesia (affecting around 1 in 10,000 people) and autosomal dominant polycystic kidney disease (affecting around 1 in 1,000 people), but they are debilitating conditions causing abnormal function of the affected areas.
The team have previously demonstrated the use of zebrafish left–right organisers – known as Kupffer’s vesicles (KVs) in fish – as an effective model organ to study renal cysts associated with autosomal dominant polycystic kidney disease. They concluded that a reduced level of Pkd2 was associated with overinflation of the model cyst caused by increased fluid secretion into the KV; this highlighted Pkd2 as an important regulator of healthy kidney function and reduced levels of the protein as a potential causal factor for cyst inflation. Nevertheless, the exact mechanism by which Pkd2 affects the development and function of these ciliated cells (including the ones in left–right organisers and renal cysts) remained unclear.
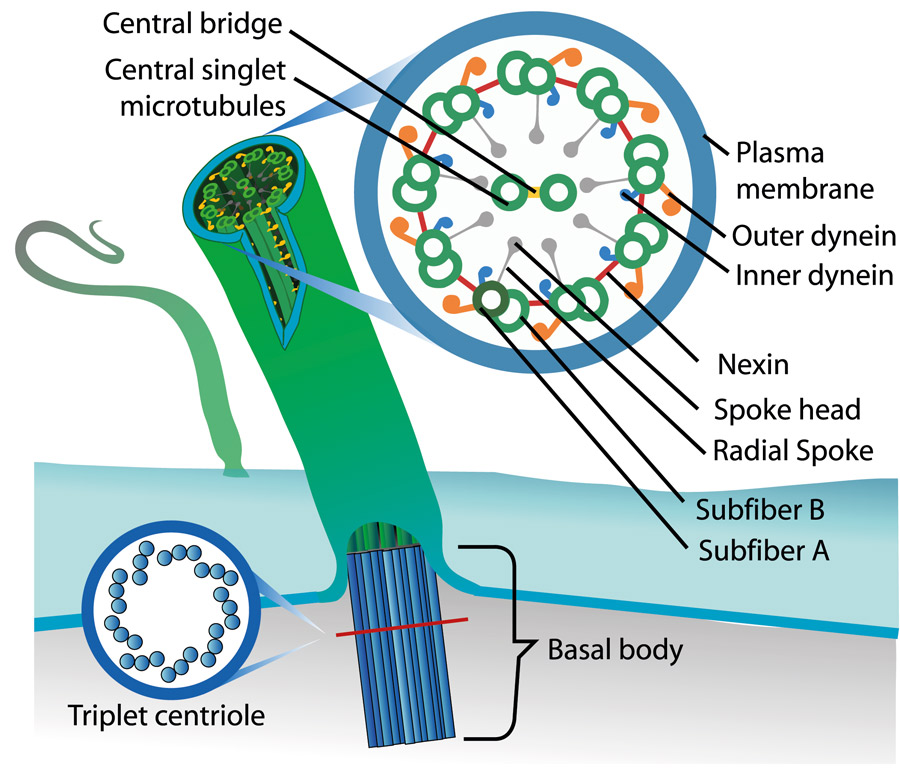
Polycystin 2 and embryonic cilia
A particular goal for Dr Lopes and her collaborators at the LYSOCIL Project was to reveal the precise activity of Pkd2 levels and their effect on the zebrafish KV. In doing so, they could establish how this protein exerts its effects on left–right development and flow dynamics, shedding light on how rare diseases might be caused.
Initially, the team investigated the KV flow characteristics in zebrafish embryos with decreased Pkd2 levels. Using antisense technology – a method of targeting genes to stop them producing certain proteins – the pkd2 gene was knocked down to produce embryonic KV cells with decreased levels of Pkd2 protein while the rest of the embryo had normal levels of Pkd2. The researchers used high-speed videomicroscopy to compare the flow speed of embryonic fluid (inside the left–right organiser) between these knocked down embryos to a control group of wild type (unchanged) embryos that had a normal concentration of Pkd2. The researchers found that the flow speed in the Pkd2-deficient embryos averaged just half that of the wild type embryos, with a rate of 5μms-1 compared to 10μms-1. This is likely attributable to the cilia beat frequency, which was also found to be lower in the pkd2 knockdown cells. The team also found a significant decrease in cell height in the affected embryos, highlighting abnormal development of the KV cells.
Dr Lopes and her colleagues next investigated the resulting length of cilia grown by the KV cells. Compared with the controls, the cilia length was shorter on average in the pkd2 knockdown embryos, with a significant decrease compared to the wild types. Interestingly, subsequent injection of pkd2 mRNA into the deficient embryonic cells caused the cilia length to increase and generated a partial rescue, though not to the level seen in the wild type cells.

The researchers wanted to take a closer look at these previously unreported findings on cilia length, to understand their implications for ciliopathic diseases. They first injected the embryos with arl13b-GFP mRNA which makes all cilia become fluorescently green, and examined the variation in the number of motile cilia of the Pkd2-deficient embryos, in which there was a reduction of more than 10% in the number of motile vs immotile cilia. Next, they re-measured cilia length in live embryos, between Pkd2-deficient mutated zebrafish KVs and nonmutated KVs. The mean cilia length was found to be 25% lower in the Pkd2-deficient cells, with an average length of 5.99 micrometres compared to 8.02 micrometres in the wild type cells expressing Pkd2 normally. The resulting decrease in flow speed is dramatic – it has been previously reported that the flow produced by a cilium is proportional to the cube of its length, so the observed decrease is expected to reduce the rate of flow by more than half.
Altered symmetry and organ positioning
The researchers finally looked to investigate the effects of reduced Pkd2 levels on the symmetry of gene expression and the resulting positioning of organs in the zebrafish. The first gene known to be expressed asymmetrically during the embryo’s development is dand5, which in zebrafish is expressed mostly on the right side of the KV. In the Pkd2-deficient embryos created using antisense technology, the resulting Pkd2 distribution was much more symmetrical, with only 22% asymmetric distribution on the right side compared to 84% in the wild type.
The findings have implications for the further understanding of rare ciliopathies, including autosomal dominant polycystic kidney disease and primary ciliary dyskinesia.
When the zebrafish embryos had developed further, the team examined their organ laterality. In the wild types, 97.5% of fish displayed situs solitus – the normal positioning of the heart and liver on the left side of the body. In the modified group, this normal organ positioning fell to 54%. Of the remaining fish, 21% exhibited situs inversus, in which the internal organs develop in the opposite orientation (heart and liver both on the right side), and 21% were heterotaxic, having the heart and liver on opposite sides. In nature, these two conditions are both extremely rare, affecting only around 1 in 10,000 human births.


Transmission electron microscopy photographs. Left: With low amplification to show a cross section through the lumen of the Kupffer’s vesicle, the zebrafish left–right organiser. Right: With large amplification to show a cross section through one motile cilia from one Kupffer’s vesicle cell.
Overall, the researchers’ results clearly demonstrate that reduced levels of Pkd2 lead to abnormal embryonic development, exhibited in misshapen cells with shorter cilia which beat less frequently. Consequently, the flow rate of embryonic fluid decreases dramatically, leading to left–right defects during growth. The findings have implications for the further understanding of rare and debilitating ciliopathies, including autosomal dominant polycystic kidney disease, as Dr Lopes and her colleagues have demonstrated that the cyst inflation question can be modelled effectively using zebrafish KVs. It also highlights questions in this field that need to be studied further – for example, are shorter cilia the cause or the effect of abnormally-shaped cells? Additional research in this area will continue to shed light on the factors behind such diseases, helping us move closer to potential treatments.
Personal Response
Your team is looking to develop a greater understanding of ciliopathies so that potential treatments may be developed – what do you think is the next major research question that needs to be addressed to make this possible?
Directly related to the Pkd2 ion channel topic, its malfunction affects at least two organs: the left–right embryonic organ and the kidneys. It is crucial to understand the role of Pkd2 in motile cilia, in non-motile cilia and in cell membranes for the laterality phenotypes of the disease primary ciliary dyskinesia, that stem from the left–right embryonic organiser. For the kidney cystic disease (ADPKD) it is important to discern the Pkd2 role in the primary cilia (non-motile present in the kidney tubules and cysts) and in the basolateral plasma membrane as well as in the ER membrane of kidney cells.