Fragment-based design of isoquinoline derivatives as anti-inflammatory drugs
Dr Masakazu Atobe from Asahi Kasei Pharma Corporation in Japan and his colleagues have developed a methodology that can be applied to the discovery of effective drugs without the need for X-ray structural information.
Modern drug discovery requires the screening of large libraries of compounds so that one effective compound can be identified as a potential drug, trialled, and eventually marketed. This approach, known as high-throughput screening, often involves the screening of hundreds of thousands of compounds a day and relies heavily on automation.
Many drugs are enzyme inhibitors – molecules that bind to enzymes, thus reducing their activity. If the target enzyme, for example, is important for the survival of a pathogen, specific drug inhibitors for that enzyme will essentially kill the pathogen, acting as antibiotics. Similarly, if the condition to treat is caused by a malfunctioning metabolic pathway, the use of an appropriate enzyme inhibitor will correct the metabolic imbalance. Since enzymes control every physiological system, enzyme inhibitors can also be used as chemotherapeutic agents, pesticides, anti-inflammatory agents or in the management of blood pressure. In order to be effective, an inhibitor needs to bind tightly to its target.
While it is crucial to test large numbers of candidate drugs, therefore increasing the probability of finding effective ones, it is even more critical that potential drugs are selected through a rational drug design process that takes into account the three-dimensional structure of the target and the chemical characteristics that make certain candidate drugs more likely to bind to it. Typically, the binding is identified from the X-ray crystal structure of the fragment bound to its target protein, enabling the rational design of candidate drug compounds. However, in many enzymatic targets, it remains difficult to design effective inhibitors, as the
X-ray structure of the ligand bound to its target enzyme is not known. Dr Masakazu Atobe from Asahi Kasei Pharma Corporation in Japan and his colleagues have overcome this problem by merging several hit fragments taken from a library of about 5,000 compounds based on the structure of isoquinoline.

Dr Atobe’s team aim to develop drugs that target a member of the Protein Kinase C (PKC) class of kinases for the treatment of rheumatoid arthritis.
Fragment merging by design
In their strategy, which the authors refer to as ‘fragment merging by design’, the fragments that successfully bind to the target are merged into one molecule to form a structure that will act as a potentially effective drug to be tested in animal models. The merging by design strategy is an adaptation of fragment-based drug discovery (FBDD), a well-established high-throughput screening method for drug generation. To date, three approved anti-cancer drugs have originated from FBDD, with several additional compounds having entered clinical trials. FBDD encompasses a broad range of techniques for finding small fragment molecules with high affinity against a target protein. Fragment molecules are typically low molecular weight, and they tend to be structurally less complex than the compounds used in conventional high-throughput screening.
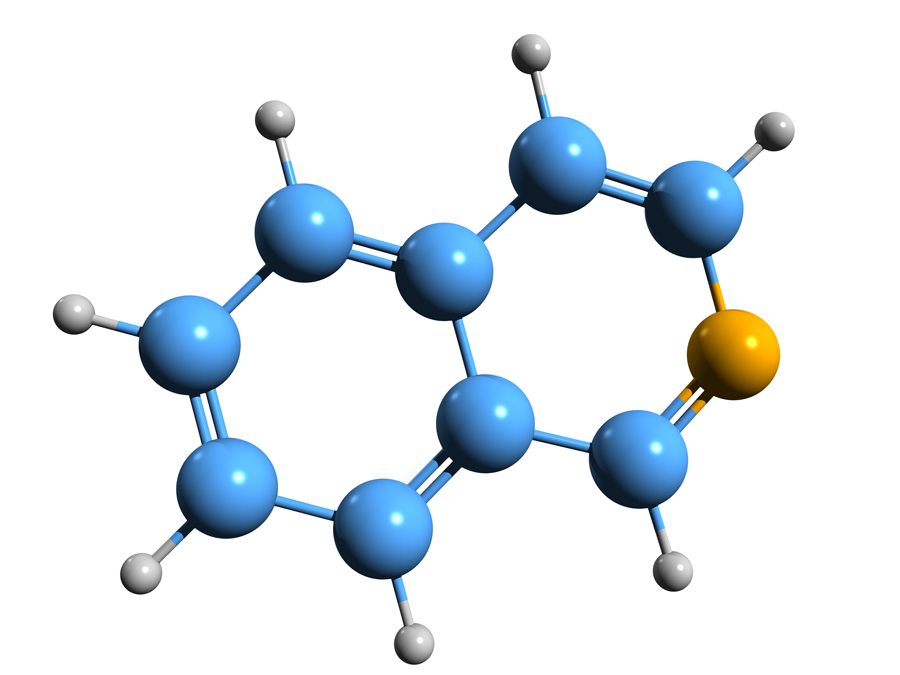
Identifying a suitable template structure
The starting point for the library built by Dr Atobe and his colleagues is the isoquinoline derivative drug fasudil, a potent inhibitor of the enzyme Rho-kinase that is commonly used as a vasodilator (medication to widen blood vessels) in the treatment of cerebral bronchospasm (a sudden constriction or ‘spasm’ of the muscles in the walls of the bronchioles) and of pulmonary hypertension (high blood pressure within the arteries of the lungs). Isoquinoline is a type of organic compound structurally known as a benzopyridine, which is composed of a benzene ring and a pyridine fused together. Isoquinoline is the parent compound of many naturally occurring isoquinoline-derivative alkaloids, which include papaverine, codeine and morphine among many others. Fasudil is known as a hinge binder for a class of enzymes known as kinases. Kinases have special binding pockets to accommodate ATP, a molecule of fundamental importance in a large number of metabolic reactions. The hydrophobic part of the isoquinoline ring occupies the ATP pocket of the kinase.

Dr Atobe’s team aim to develop drugs that target a member of the Protein Kinase C (PKC) class of kinases for the treatment of rheumatoid arthritis. PKC inhibitors are also believed to be ideal candidates in the treatment of numerous conditions, such as diabetes, chronic inflammation of the skin, and breast cancer.
Analysis and optimisation of the hit fragments
To generate suitable fragments, the team at Dr Atobe’s laboratory synthesised several chemical structures that would each bind one among the 1-, 3-, 4-, 5-, 6-, 7-, or 8-positions of the isoquinoline ring to produce the library of 5,000 isoquinoline derivatives. They anticipated that the biochemical screening of monosubstituted isoquinoline fragments would provide many hit fragments. Once two fragments substituted on different positions were identified, they were merged on the same isoquinoline ring, rapidly providing a highly potent molecule without the need for previously obtained structural information.
The ‘merging by design’ strategy can be applied when the X-ray crystal structure of the fragment bound to its protein
is not known.
In their latest paper, two examples of fragment merging by design were provided for two kinase inhibitors. They first merged the derivative substituted at position 4 of the isoquinoline ring with one substituted at position 6, resulting in a compound that yielded mid-nanomolar inhibition of its target kinase. Next, they merged the 5-substituted isoquinoline derivative with a 7-substituted derivative to generate a very potent compound, capable of acting as a kinase inhibitor at less than nanomolar concentrations. Further optimisation led to the 5,7-disubstituted isoquinoline, which was subsequently tested on an animal model study of rheumatoid arthritis, resulting in good efficacy.

Concluding remarks
The ‘merging by design’ methodology is a promising pharmacological strategy that can be applied to the discovery of challenging kinase inhibitors when the X-ray crystal structure of the fragment bound to its protein is not known. The technique is an adaptation of FBDD and can be summarised as a five-step process. The first step consists of a biochemical screening of fragments that belong to a common template structure. In the work of Dr Atobe and his colleagues, isoquinoline proved to be a good template for the development of kinase inhibitors and a library of 5,000 fragments was initially screened. The second step is the analysis of the fragments that produced a ‘hit’, in other words a successful binding event with the target protein. In the case of isoquinoline, the fragments can originate from derivatives where the original double-ring-molecule is attached to a chemical substituent that can replace hydrogen on any one of the seven available positions on the double ring. The third step, fragment merging, is key to the design strategy. When two fragments originate from substitution on two different positions of the template structure, they can be merged, rapidly providing a highly potent molecule. Through this strategy, Dr Atobe and his colleagues managed to narrow down the search from the original 5,000 fragment compounds to the rapid identification of two chemical series of disubstituted isoquinolines. Once the potential drug candidates are identified, the fourth step, optimisation, consists in their synthesis and validation, conducted by assessing their effectiveness in cell culture studies. Finally, the most potent and orally available compounds are selected for more studies on animal models. Further in vitro analysis of a 5,7-disubstituted isoquinoline inhibitor proved to the team that it was a good candidate drug for the treatment of rheumatoid arthritis. The in vivo studies in a model of the disease in mice confirmed that the compound is effective in treating rheumatoid arthritis, acting as an anti-inflammatory with disease-modifying properties. In future, the team hopes to use the compound as a chemical tool to explore further the efficacy, safety and tolerability of inhibitors of the protein kinase C class of enzymes.

Personal Response
Will any of the compounds you identified be undergoing clinical trials in the near future?
What are you looking to research next?