Disruption of amyloid β-protein processing drives Alzheimer’s
In the study of familial Alzheimer’s disease, a specific form of amyloid β-protein (Aβ) found in amyloid plaques has been the focus of many investigations as a key driver of disease. However, increased concentrations of this form of Aβ does not always occur with disease-causing mutations, suggesting a different step in Aβ processing could be triggering Alzheimer’s. Professor Michael S. Wolfe and his team at the University of Kansas explored this problem further and determined that processive cleaving by γ-secretase was consistently reduced in familial Alzheimer’s disease cases.
Alzheimer’s disease is a devastating and common condition that causes memory loss and impaired cognitive function. Studies to understand the molecular basis of pathological features of Alzheimer’s – including cerebral neurodegeneration, build-up of extracellular amyloid plaques, and intraneuronal neurofibrillary tangles – have been ongoing for decades. Deciphering the causes of these pathologies has been challenging, due to the complexity of the disease. No effective treatments have been developed for Alzheimer’s, in part due to incomplete understanding of the underlying drivers of the disease process.
A rare inherited form of the disease, familial Alzheimer’s disease, presents with similar symptoms and pathology to the more common sporadic Alzheimer’s disease. The only clear differences are that familial Alzheimer’s occurs in midlife rather than old age and is caused by single dominantly inherited mutations. These mutations are found in genes encoding proteins directly responsible for the production of a sticky protein that builds up in the amyloid plaques. The dominant inheritance, early age of onset and connection to amyloid pathology makes familial Alzheimer’s easier to study than the common form of the disease in the elderly. Research into familial Alzheimer’s disease is important not only for carriers of these devastating mutations but for all Alzheimer’s patients due to the similarity between these two forms of the disease. Finding new targets and strategies for developing treatments is essential for improving quality of life for patients and their families. While some targets are already known, not all drivers of Alzheimer’s pathology have been discovered.

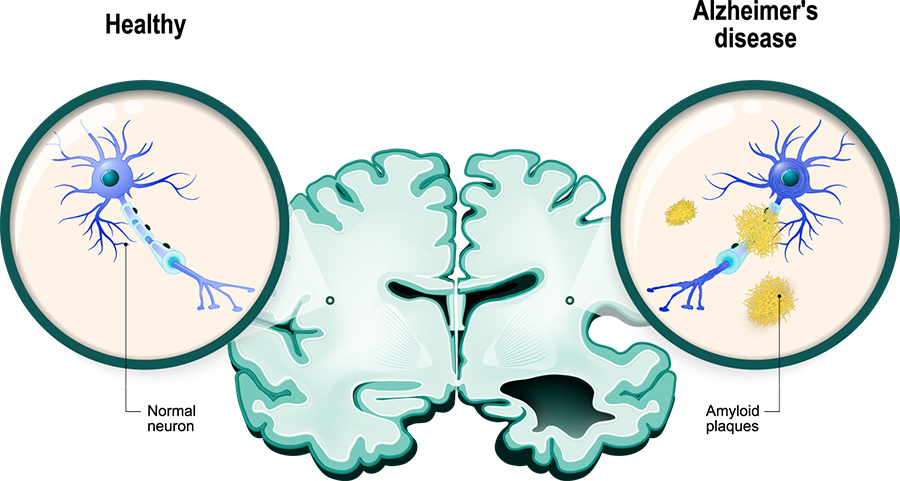
Amyloid plaques
One of the key components in the development of the disease is the presence of cerebral amyloid plaques. These plaques are outside the nerve cells in the brain and are made from a build-up of the amyloid β-protein (Aβ). This protein is formed when an amyloid β-protein precursor (APP) is cleaved by a protease enzyme called γ-secretase. The Aβ produced can be of varying lengths, but only proteins of 43 amino acids or fewer in length are secreted from the cells. Aβ that is 42 amino acids in length (Aβ42) is the primary form that builds up in the plaques.
These results show that deficient processing of long Aβ by γ-secretase is a common feature of mutations that cause familial Alzheimer’s disease
In patients with familial Alzheimer’s disease, 3 genes have been discovered to have inherited dominant missense mutations (mistakes within the DNA leading to incorporation of an incorrect amino acid). These are APP and presenilins-1 and -2, which are γ-secretase components. Mutations in these genes can cause proportionally more Aβ42 to be produced, leading to development of amyloid plaques. For this reason, Aβ42 and plaque formation is considered integral to triggering the disease process. However, treatments targeting Aβ42 have not shown effectiveness in slowing or halting the decline of Alzheimer’s disease, and not all mutations associated with familial Alzheimer’s disease lead to elevated proportions of Aβ42, suggesting there is more to this process.
Processive proteolysis
To more fully understand the factors that could be affected in familial Alzheimer’s disease, researchers should investigate the activity of the γ-secretase enzyme more closely. In cleaving the APP, the enzyme does not catalyse only a single cut but also performs a series of processive trimming events. The first cut is carried out at the cytosolic end of the transmembrane domain of the APP. This cut causes the intracellular domain of the APP to be released into the cytosol while either Aβ48 or Aβ49 (48 or 49 amino acids in length, respectively) remain membrane bound.
The γ-secretase enzyme then continues trimming the membrane-bound protein in increments of 3 amino acids. This gives two different pathways of processive proteolysis: Aβ49 → Aβ46 → Aβ43 → Aβ40 and Aβ48 → Aβ45 → Aβ42 → Aβ38 (this last cut is 4 amino acids long). Both pathways result in a series of short peptide chains 3 (or 4) amino acids along with the shorter Aβ proteins being produced. The shorter proteins are secreted once the cuts remove most of the transmembrane domain and the Aβ protein is no longer bound to the membrane. This is what usually occurs when no mutations are present, but the mutations associated with familial Alzheimer’s disease could also be affecting this part of the process.
Mutants affecting trimming steps
In order to better understand effects of mutations that cause familial Alzheimer’s disease on this series of proteolysis trimming events, Professor Michael S. Wolfe and his team at the University of Kansas decided to investigate this further. The first experiments they carried out analysed the ratios of Aβ42 to Aβ40 in 14 disease-causing mutations in APP compared to the wild-type protein. The mutants were all missense mutations within the transmembrane domain of APP, in or around γ-secretase cleavage sites. The results showed that 3 of these mutants had no increase in the ratio of Aβ42 relative to Aβ40, confirming again that Aβ42 is not the only pathological driver of the disease, as Alzheimer’s can develop without changing its proportion. This further highlights the need to better understand the effects of disease-causing mutations on the other cleavage steps and aspects of the proteolytic process.

Analysing each step of the process quantitatively presented some difficulties, as the more usual methods of testing were not possible. The longer Aβ proteins were too prone to aggregation to use mass spectrometry and had no specific antibodies for a common colour-based assay system called ELISA. The team therefore decided instead to measure levels of the very short peptides of 3-4 amino acids produced in the trimming stage of the process. From that information, the level of each form of the Aβ protein present can be inferred and determined. These experiments used liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS). The liquid chromatography allows separation of the different small peptides, while the mass spectrometry leads to identification and quantification of each peptide. These experiments showed that the trimming stages were slower with the disease-causing mutant forms of APP. The mutants all showed a decrease in the first and/or second trimming step in the Aβ40 and/or Aβ42 pathways compared to the wild type. This means that more of the longer Aβ proteins remain, instead of undergoing the expected processive proteolysis.

These longer Aβ proteins remain bound to the membrane and may be a key driver of disease progression.
The overall levels of different Aβ peptides were calculated using this information as well as results from analysis of the APP intracellular domain, which informs whether the first proteolytic event results in Aβ48 or Aβ49. The net levels of these Aβ peptides were determined by subtracting the amount of small peptide degradation from the amount of small peptide production, giving a complete and quantitative view of each pathway. These calculations revealed that all the mutants gave overall increased levels of longer Aβ proteins between 45 and 49 amino acid residues in length. This is due to reduced efficiency in γ-secretase during the first and/or second Aβ trimming steps, leading to less of the shorter proteins. These longer Aβ proteins remain bound to the membrane, as they contain most of the transmembrane domain, unlike the shorter ones which are secreted. This discovery is a key difference from the wild-type process, common to all 14 of the disease-causing APP mutations that were studied. Further research into understanding the normal or pathological effects of these membrane-bound Aβ proteins is needed.

Future therapies
The importance of understanding the pathological causes of Alzheimer’s disease cannot be overlooked. The experiments undertaken by Michael Wolfe and his team have shed some light on the key process of APP proteolysis by γ-secretase. The discovery that these longer membrane-bound Aβ proteins are overproduced when not cleaved correctly, as a result of disease-causing mutations, could lead to a new line of investigation. The focus of Alzheimer research has primarily been on Aβ42 and its role in amyloid plaque build-up and pathology, but this needs to change, as Aβ42 is clearly not the only cause of the disease. More work needs to be done to understand the role of these long forms of Aβ and how they may be driving familial Alzheimer’s disease. These results clearly show that the ineffective processing of Aβ by γ-secretase is key for disease progression. This deficient processing presents a potential target for future therapies that could provide effective treatment for patients suffering across the globe.
Personal Response
What do you think is the next step in investigating these Aβ that are not secreted?
Key experiments should explore the potential role of these long Aβ peptides and the deficient γ-secretase trimming process in disrupting brain nerve cells and their connections. We are testing effects of Alzheimer-causing mutations in cell and animal models under conditions in which Aβ42 is not produced. Ruling out Aβ42 as responsible for disruption of nerve cells would clearly point to a critical role of deficient trimming of long Aβ peptides in triggering familial Alzheimer’s disease. Such findings should have implications for the aetiology and treatment of the much more common Alzheimer’s disease in those over 65 years of age.