Improving the effectiveness of an anticancer fluoropyrimidine by molecular hybridisation
Capecitabine (Cape) – a fluoropyrimidine used primarily in the treatment of breast and colorectal cancer – disrupts the synthesis of DNA in cancer cells following its conversion to 5-fluorouracil (FU). Since FU has been associated with several potentially fatal side effects, Dr Thomas I. Kalman at the University at Buffalo and The State University of New York has developed a derivative of Cape that is not metabolised to FU, while at the same time enhancing its anticancer activity. The novel compound, a hybrid drug known as pencitabine (Pen), is the result of the fusion between Cape and gemcitabine (Gem), another anticancer drug.
Capecitabine (Cape) is an orally active anticancer drug belonging to the class of compounds known as fluoropyrimidines, which are used in the treatment of colorectal and breast cancer. Cape is a pro-drug: an inactive compound that is metabolised by the body and converted to the active drug 5-fluorouracil (FU), which exerts a potent chemotherapeutic action by blocking the DNA synthesis in malignant cells. There is, however, a very narrow window between the minimum effective dose of Cape and the maximum tolerated dose of its metabolite FU, which is associated to a multitude of potentially fatal toxic side effects and damages normal cells as well as cancerous ones. Metabolites of FU have been implicated in neurotoxicity and likely cause several other toxic side effects.
Two strategically placed fluorine atoms in Pen’s structure prevent its conversion to 5-fluorouracil and its associated potentially fatal toxicities.
Dr Thomas I. Kalman, Professor Emeritus at the University at Buffalo and The State University of New York in the US, redesigned the structure of Cape by drawing on his expertise in medicinal chemistry of enzyme inhibitors. He has modified the structure of Cape to prevent its metabolic conversion to FU. In an article published in 2022, Kalman reported the rational design, synthesis, and biological activity of Pencitabine (Pen), a novel, orally active derivative of Cape that cannot be metabolised to FU. The preliminary investigations described in his study suggest that Pen retains the chemotherapeutic action of Cape but is devoid of FU-related toxic side effects.

Overcoming therapeutic resistance
FU, Cape’s active metabolite, acts by inhibiting the enzyme thymidylate synthase (TS) in cancer cells. Enzymes are biological molecules that act as natural catalysts, facilitating and speeding up chemical reactions in biological processes. TS is a pivotal enzyme in the synthesis of DNA, therefore its inhibition will in turn block cell division and cancer growth. Pen is a hybrid drug, synthesised by the fusion between Cape and another anticancer drug known as gemcitabine (Gem). The hybrid drug is orally active and shares many of Cape’s structural features, but the addition of two strategically placed fluorine atoms in Pen’s structure prevents its conversion to FU and its associated potentially fatal toxicities.

Similarly to its parent drugs, Pen halts the DNA synthesis in cancer cells. However, Pen also blocks the DNA damage repair mechanisms of cancer cells, which means that the drug can potentially overcome chemotherapeutic resistance. The main advantage of incorporating two drugs into one molecule is that they can act simultaneously. In his 2022 publication, Kalman reports that clinical trials of the combination of oral capecitabine and intravenous (IV) gemcitabine for the treatment of pancreatic cancer have confirmed an advantage over treatment with either of the two drugs alone. The hybridisation of Cape and Gem into Pen eliminates the requirement for IV administration of the gemcitabine component while maintaining its therapeutic contribution in oral form.
Experiments in mice confirmed that when Pen was given orally at a dose of 25mg/kg, it inhibited 48% of the growth of human colon cancer tissue that was artificially implanted in rodents. It was shown that, to achieve the same degree of growth inhibition, it was necessary to use an amount of Cape that was equivalent to four times the dose of Pen used in the experiments.
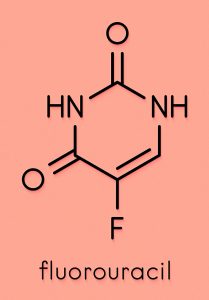

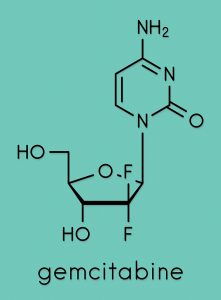
Despite the preliminary nature of the investigations undertaken, Kalman is confident that more experiments will confirm that Pen has a significantly higher therapeutic power than Cape. In cell culture, Pen was found to work against both solid tumours and leukaemia (a group of blood cancers). Kalman also noted that modification of the structure of Cape increased its potency, significantly lowering the concentration at which the drug exerts its effect on cancer cells. Although a detailed investigation of the mechanisms of action of Pen has not been conducted, reasonable predictions of its pharmacological targets can be made based on the studies already carried out on both Cape and Gem.

Comparing the mechanism of action
Cape and Pen act in a similar way. Both provide an example of mechanism-based inhibition, a form of enzyme inactivation that is achieved by a diverse range of therapeutics, including aspirin and penicillin among many others. Mechanism-based inhibitors are chemically unreactive in their native form, but they are activated by their target enzyme, forming a covalent bond with the enzyme itself and causing its inactivation. Pen’s intracellular metabolites can inactivate three of its target enzymes by mechanism-based inhibition: ribonucleotide reductase, thymidylate synthase and DNA (cytosine-5)-methyltransferase.
While the primary enzyme target of Cape is the above-mentioned TS, Pen also acts on a different enzyme, DNA methyltransferase (MT). Kalman proposes that, in addition to its effect on DNA structure and function, Pen may also cause inhibition of DNA glycosylase enzymes involved in DNA repair mechanism. Another important difference is that, while the inhibition of TS by Cape is due to the formation of a bond that can be reversed, the inhibition of MT by Pen happens via the formation of an irreversible chemical bond.
Malignant tumour in the intestines (left) and microscopic photograph of colon cancer (right).
Kalman explains that the presence of three fluorines at strategic positions in the structure of Pen is responsible for its different and improved anticancer properties when compared with Cape. It is true, however, that Kalman’s hypothesis on Pen’s mechanism of action needs to be validated through further experimentation in future studies. According to Kalman, due to its multiple DNA-targeted effects, Pen may also reduce the likelihood of the emergence of drug resistance and can be used in combination with other DNA damaging agents, such as radiation and platinum-based drugs, to achieve greater therapeutic efficacy.
Due to its multiple DNA-targeted effects, Pen may also reduce the likelihood of the emergence of drug resistance.
Concluding remarks
Kalman developed a new, orally active anticancer drug, Pencitabine, by combining the structures of Cape and Gem, two chemotherapeutics belonging to the class of compounds known as antimetabolites. The researcher redesigned the structure of Cape to prevent its metabolic conversion to 5-FU, which is associated with potentially fatal toxicity. The product retained the beneficial effects of the therapeutic combinations of Cape and Gem. Preliminary studies showed that Pen causes inhibition of two enzymes of DNA metabolism and that neither of the two components of the hybrid, Cape or Gem, alone were reported to inhibit these enzymes. Since Pen has an increased number of targets and one of the two target enzymes is involved in DNA repair mechanism, it is possible that the hybrid drug could be effective in overcoming chemotherapeutic resistance in cancer cells. Kalman hopes that the results from his preliminary studies will be corroborated in further experimentation, so that the mechanism of action of Pen can be thoroughly validated and its safety and efficacy confirmed.

Personal Response
What further steps need to be followed to confirm the therapeutic potential of Pencitabine?
Further development of Pencitabine as a drug candidate will require complete preclinical pharmacology and toxicology studies followed by clinical trials. Approval by the Food and Drug Administration (FDA) must await the favourable results of these studies.