How oxidative stress contributes to neurodegenerative diseases
The human brain relies on a constant energy supply that when compromised, neurodegenerative disease ensues, initially disrupting nerve cell communication followed by nerve cell death. Paul Hyslop and Michael Chaney investigate fundamental mechanisms driving neurodegenerative disease, to identify therapeutic interventions. They focus on how an enzyme integral to energy supply to nerve cells, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) – is inhibited and transformed to a toxic complex by hydrogen peroxide (H2O2) – a product of brain cell activity that becomes elevated in neurodegenerative diseases. Their work provides important clues to the factors affecting nerve cell health and offers potential therapeutic avenues for treatment of neurodegenerative disease.
The brain is a complex and fascinating organ. It is responsible for processing signals generated by the five senses, integrating and interpreting the information they carry, and creating, quickly and efficiently, suitable bodily responses to changing environmental conditions. It also supports the highest functions of which the human mind is capable, including thought, emotions, and language.
The brain is composed of roughly 86 billion nerve cells called neurones, which communicate with each other by exchanging chemical and electrical signals. Neurones cells are part of a formidably intricate communication network, whose structure evolves over time to accommodate new experiences in the form of new connections among subsets of neurones.
Brain health and energy supply
To guarantee a continuous supply of energy to neurones and the removal of dangerous chemical waste, the human body has evolved complex networks of hugely cooperative mechanisms. Specialised non-nerve cells, known as glial cells, constantly interact with the surface of the neurone. They provide nutrients, remove waste and toxins, and regulate the chemical environment of the brain. When any of these functions fail to support neurone activity, their structural integrity is compromised and ability to communicate with each other reduced. This can lead to neuronal death, and reduction in the overall connectivity between neurones, as most neurones in the brain cannot regenerate.
Neurodegenerative diseases
Although neurones naturally and slowly die with age, pathological conditions can accelerate this process. This results in mental confusion, slowing of everyday thinking, and other symptoms typically ascribed to a class of mental conditions collectively known as dementias, of which Alzheimer’s and Parkinson’s diseases are well known examples. The symptomatic complexity of dementias and the highly heterogeneous distributions in their degree of severity and rate of mental decline pose enormous challenges in the development of effective treatment approaches.
Bacteriostasis
In higher life forms, a component of the defence mechanism against the killing of external pathogens is mediated in part by the production of reactive oxygen species. H2O2 acts as very efficient chemical weapons, halting growth of invading microorganisms, a phenomenon known as bacteriostasis [1]. However, to prevent damage to the host cells, including the brain’s neurones, the concentration of these molecules must be carefully regulated. Paul Hyslop and colleagues at Arkley BioTek LLC, Indianapolis, believe this could be key to understanding how neurodegenerative conditions arise, evolve over time, and how they might offer an avenue for treatment.
…decreasing chemical energy supply and effective elimination of toxic waste products act synergistically to define the point at which a critical threshold for neurodegeneration is crossed.Oxidative stress and neurodegeneration
‘Our research premise for therapeutic development,’ explain Hyslop and Chaney, ‘is that decreasing chemical energy supply and effective elimination of toxic waste products act synergistically to define the point at which a critical threshold for neurodegeneration is crossed.’ Their work is specifically devoted to the fine details of how a key enzyme involved in metabolism, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), interacts with H2O2 .
GAPDH plays a crucial role in glycolysis, the metabolic pathway that produces energy by metabolising glucose molecules to generate adenosine triphosphate (ATP) molecules, the main carrier of energy in cells.
GAPDH is widely distributed in tissues and, in addition to its metabolic function, it plays a role in other vital tasks, including DNA repair, regulation of gene expression, and controlled cell death, or apoptosis. Hyslop and Chaney’s findings contribute to the growing body of scientific literature demonstrating that oxidant modification of GAPDH by H2O2 contributes to neurodegenerative diseases.
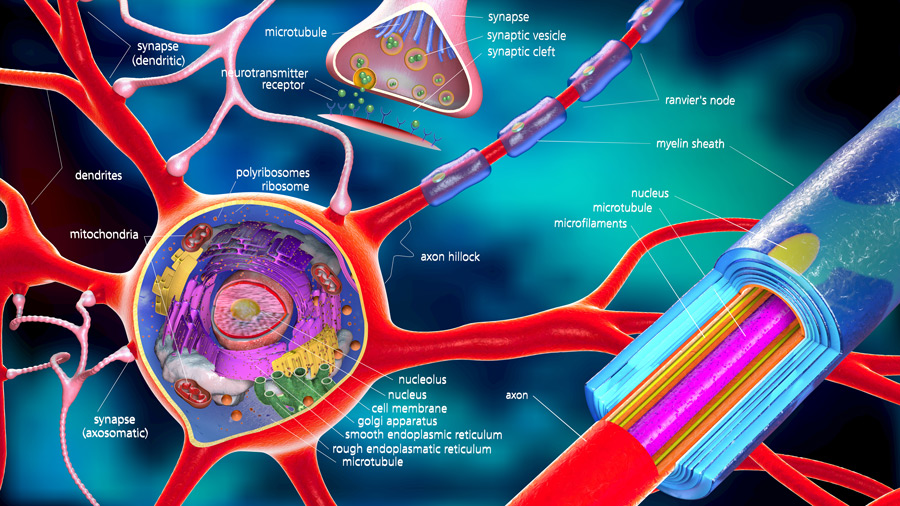
A closer look at GAPDH
All enzymes are polymeric chains of amino acids, arranged according to a specific sequence known as primary structure. Through electrostatic and covalent bonding, these chains acquire a characteristic three-dimensional shape, which includes secondary structures, such as alpha helices and beta sheets. The overall three-dimensional shape is known as the tertiary structure. Proteins can also form complexes with other protein chains (quaternary structure). The cellular environment and selective modifications influence the tertiary structure of each component. These modifications enable interactions with other proteins, promoting essential communication and signalling functions within and between cells.
GAPDH is a tetramer, a group of four identical subunits each of which contains one active site – where the enzyme reaction takes place. If the ability of GAPDH to carry out this chemical reaction is compromised, the cell looses its functionality.
Cell signalling
When a neurone is under stress, GAPDH can undergo chemical modifications, such as oxidation and nitrosylation, which shift its physiological function from a metabolic enzyme to a signalling molecule. These modifications promote the interaction of GAPDH with chaperone proteins, which assist its translocation from the cytoplasm to key intracellular organelles such as the nucleus [2] and mitochondria [3]. The nucleus is responsible for orchestrating maintenance and repair of cell structures and function, while mitochondria produce the cells energy supply in the form of ATP. Within the mitochondria and nucleus, translocated GAPDH (in addition to other important parallel signalling complexes), results in cellular stress response dictating cell fate decisions, either promoting neuronal survival or initiation of ‘programmed cell death’, referred to as apoptosis. As we age, apoptosis slowly diminishes our complement of neurones.
However, when neurodegenerative disease (such as Alzheimer’s disease) afflicts an individual, the rate of neuronal apoptosis accelerates uncontrollably with tragic consequences. At present, the molecular signals that trigger neurodegeneration are poorly understood, and is the subject of intense research into identifying probable causes at the molecular level to target with novel pharmaceuticals. Although translocation of oxidised GAPDH to cellular organelles is known to play a crucial role in apoptosis, we do not know to what extent dysregulation of this process contributes to neurodegenerative diseases. To address this question, it is first necessary to study and understand how GAPDH is conformationally modified by oxidation to trigger binding to its molecular chaperones.
GAPDH oxidation
Using a combination of experimental techniques and computer simulations, Hyslop and Chaney have investigated the details of the mechanism through which H2O2 influences both the ability of GAPDH to supply energy to neurons via the metabolism of glucose, and to bind to signalling chaperone proteins. Their approach starts from careful biochemical measurements resulting from chemical modifications following H2O2 oxidation of commercially purified GAPDH, and then applying these modifications directly to the native GAPDH crystal structure. The computation then calculates the new ‘energy minimised’ secondary and tertiary structure resulting directly from the oxidative modifications.
These findings provide novel insight into the biochemical phenomena leading to neurodegeneration in the brain.Hyslop’s and Chaney’s first analysis shows that first, H2O2 oxidises the catalytic cysteine residue, and then sequentially oxidises a second neighbouring cysteine group. The oxidised cysteines react with each other, forming an interchain thiosulfinic ester bond – causing a major rearrangement in the structure of the enzyme’s subunits, predicted to induce a sufficient change in the binding affinity of GAPDH to chaperone proteins involved in the translocation of the complex to the nucleus and mitochondria and activation of apoptosis. Based on the work of others, oxidation of GAPDH and its translocation to the mitochondria also promotes the production of H2O2, further increasing the level of cellular oxidative stress.
Antioxidants
Cells use antioxidants to counteract the destructive effects of reactive oxygen species. Glutathione is one such molecule that can protect the GAPDH catalytic cysteine residue from oxidation by H2O2 , forming S-glutathionylated GAPDH. The process is reversible, allowing fully functional GAPDH to be regenerated with the help of a second glutathione molecule directly or with the help of glutathione transferase enzymes. After H2O2 oxidation of the catalytic cysteine residue, oxidation of the vicinal cysteine residue is prevented by S-glutathionylation, inhibiting the formation of the GAPDH/chaperone signalling complex.


Figure 1. Cartoon representation of GAPDH enzyme subunit using the crystal structures modified by both H2O2 oxidation and S–glutathionylation.
(A) Native GAPDH and H2O2-oxidised GAPDH superposed subunit structures of GAPDH demonstrates transposition of the ‘S-loop’ region after the subunit enzyme. The S-loop region is a potential site for binding of oxidised GAPDH with other cellular proteins to form complexes involved in cell signalling events. (B) The same analysis was performed as in panel (A), except that the native subunit was superposed with S-glutathionylated subunit. Importantly, there are clear differences between the structural features between the H2O2-oxidised and S-glutathionylated structures, indicating that the two modifications to GAPDH potentially interact with different cellular proteins that mediate different signalling events.
In both cardiovascular and neurodegenerative diseases S-glutathionylated GAPDH has been shown to accumulate in tissue and blood. It is also known that following either ischaemic injury or oxidative stress to isolated cells, GAPDH function is inhibited and can account for the inhibition of metabolic energy available to the cell from glucose. Hyslop and collaborators were puzzled by this observation, and how it might tie into the role of GAPDH as an integral participant in both providing cells with metabolic energy, available to the cell from glucose, and its role in apoptosis. In a recently published companion study, they discovered important clues to this question. When the active site cysteine of GAPDH is S-glutathionylated, S-glutathione binds tightly with neighbouring amino acids within the active site pocket, effectively inhibiting its removal and reactivation of and its role in apoptosis, providing insights to its role in inhibition of glucose metabolism and its accumulation in cardiovascular and neurodegenerative diseases.
Using the same techniques of molecular dynamic simulation, the energy minimised structure of S-glutathionylated GAPDH shows that H2O2 oxidation of both the catalytic and neighbouring cysteines (glycolysis block and apoptosis) and S-glutathionylation of the H2O2 oxidised catalytic cysteine (glycolysis block) affect the structure of GAPDH in subtly different ways. A specific, relatively flexible sequence of amino acid residues within GAPDH subunits known as the ‘S-loop’, (amino residues alanine 180–leucine 203) was shown to be significantly conformationally altered as a consequence of H2O2 oxidation of the two cysteine residues, and also by H2O2 oxidation of the catalytic cysteine residue followed by its S-glutathionylation compared to native GAPDH subunits (figure 1).
However, the final structure of the two modified GAPDH subunits are significantly different in the two cases. A superposition of the structure of a subunits of native and H2O2 oxidised GAPDH (figure 1A) and native and S-glutathionylated GAPDH (figure 1B) with the native enzyme structure shows substantial differences in the orientation of the S-loop. As a visual guide the colour-coded tryptophane 196 residue side-chain is added to the subunit backbone to illustrate change in the overall conformation of the protein. The S-loop is thought to be a potential binding site for cellular proteins involved in cell signalling events, and, according to Hyslop and Chaney’s findings, the two different structures could bind to different signalling proteins. On one hand, only in the case of GAPDH with both catalytic and neighbouring cysteines oxidised, do these binding events lead to apoptosis. S-glutathionylated GAPDH on the other hand could signal to the cell nucleus that a condition of oxidative stress exists, and countermeasures to promote neuronal survival are activated. The conclusions of these findings are that GAPDH is key to understanding the intersection of oxidative stress, energy metabolism, and apoptosis and how cell fate decisions are guided.
From biochemistry to therapy
These findings provide novel insight into the biochemical phenomena leading to neurodegeneration in the brain. The use of computer simulations also makes it possible to study in silico how chemical modification of GAPDH can increase its resistance to oxidative stress. ‘This work,’ explains Hyslop and Chaney ‘should facilitate a greater understanding of signalling pathways involved in cell fate decisions and assist in the identification of new targets for therapeutic intervention, particularly for chronic neurodegenerative diseases.’
Personal Response
Building from your results, what are the most promising future approaches for the treatment of neurodegenerative conditions induced by oxidative stress?It has been important to understand exactly how H2O2 and glutathione interact with GAPDH in order to identify sufficiently stable conformers to generate crystals for x-ray analysis of their structure. This will result in being able to visualise the exact conformational changes predicted by computational modelling analysis. At the same time, these sufficiently stable conformers of GAPDH will be used to bind to cell extracts to identify novel binding protein complexes. Once identified, the docking sites between the two proteins can be evaluated using computational methods. Our goal will be to identify specific protein interactions that drive neurodegeneration following oxidative stress, and to begin to design targeted drug compounds directed to inhibit neuronal cell death.