Regulators of protein degradation as potential treatments for neurodegenerative disease
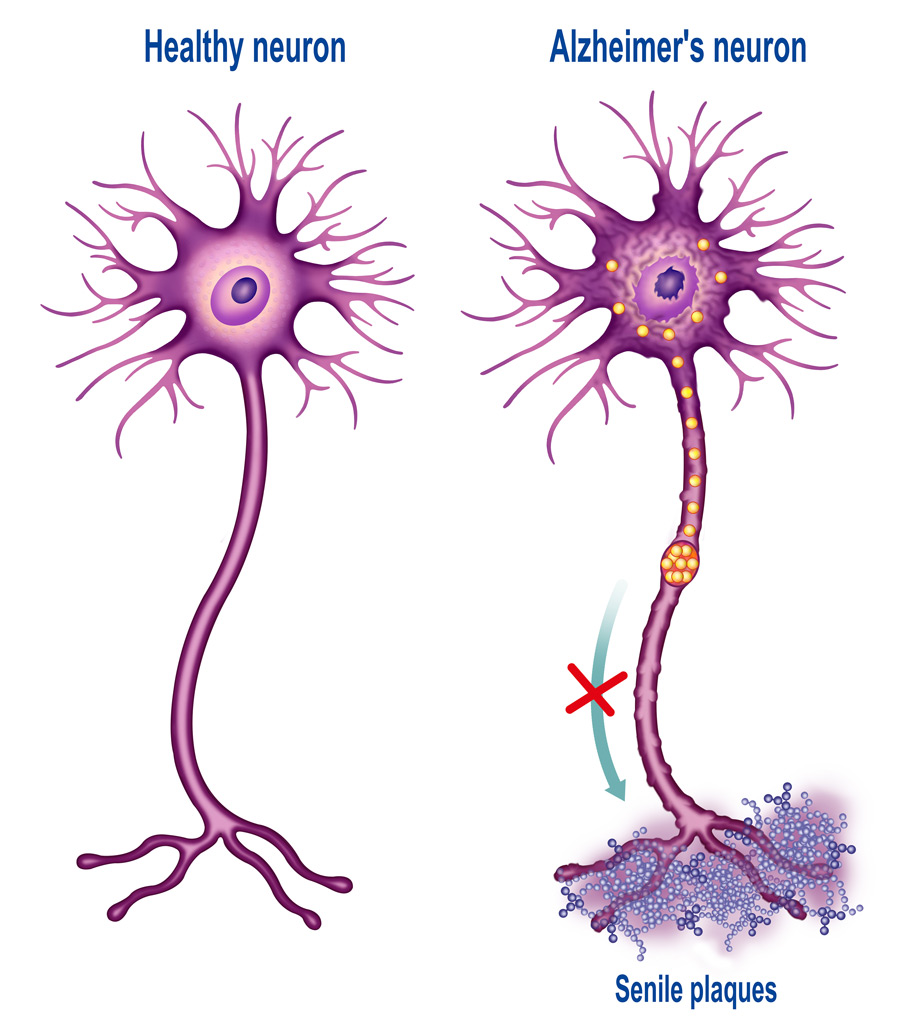
Neurodegenerative diseases are characterised by the progressive impairment of the normal structure and function of neurons, ultimately leading to their death. Since neurons aren’t normally replaced after their death, the damage to the central and peripheral nervous system is irreversible and results in the severe impairment of movement or mental functioning. Millions of people around the globe are affected by neurodegenerative diseases and the risk of developing neurodegeneration increases with age, meaning that with more people living longer worldwide, the need for a breakthrough in the development of effective treatments is more urgent than it has ever been.

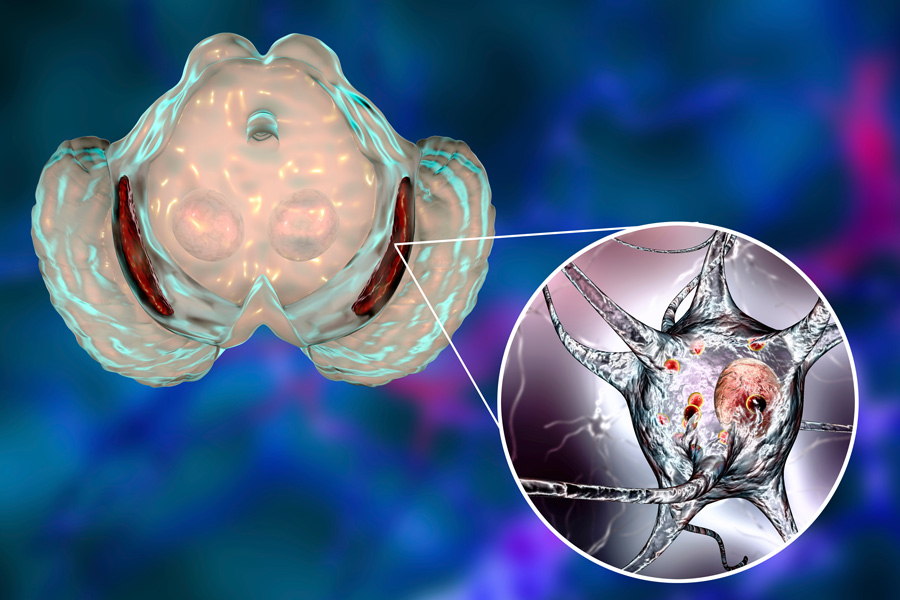
Cells have structures that act as ‘quality check’ mechanisms, ensuring that proteins that are damaged or malfunctioning are flagged for destruction. The main cellular apparatus responsible for protein degradation, the proteasome, is a marvellous example of molecular machinery. The proteasome itself is impaired in several instances of neurodegenerative diseases. Despite many studies conducted on the proteasome, an impairment mechanism in neurodegenerative diseases has not been identified yet.
Their findings suggest that there could be a key mechanism of proteasome impairment that is common to several neurodegenerative diseases.
Dr David Smith and his team at the School of Medicine, University of West Virginia, have many years of experience in the research of the molecular mechanisms behind proteasome function. They have recently focused their efforts on understanding how the accumulation of neurotoxin protein aggregates can inhibit or impair the proteasome in many of the most common neurodegenerative diseases. In this context, they have investigated the hypothesis that there exists a universal mechanism of proteasomal impairment, which is common to the major neurodegenerative diseases, such as Alzheimer’s, Huntington’s and Parkinson’s disease. In 2018, they published a study reporting that specific small soluble oligomers of three disease-related proteins (beta amyloid, alpha-synuclein, and polyQ expanded huntingtin) that share a common misfolded morphology, act as potent inhibitors of proteasome function. Their findings suggest that there could be a key mechanism of proteasome impairment that is common to several neurodegenerative diseases.

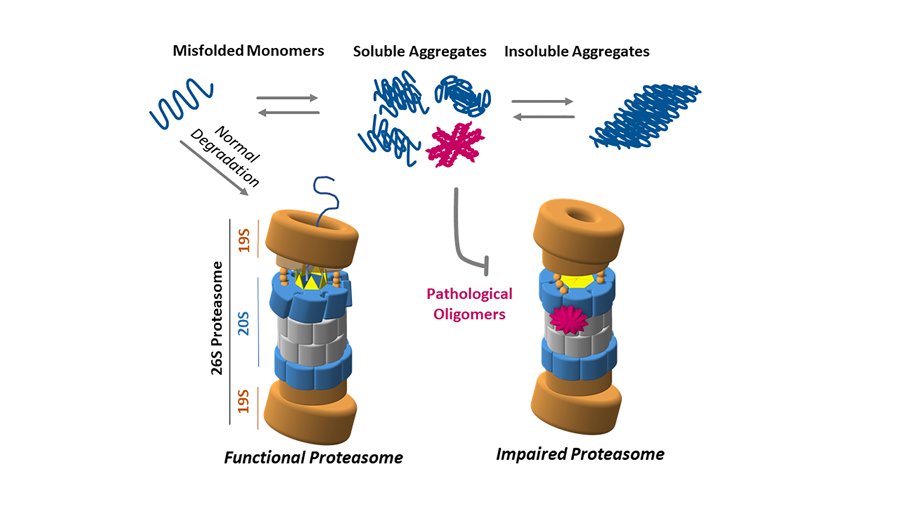
Small pathogenic oligomers escaping degradation
A vast amount of evidence suggests that large aggregates of proteins are not per se the cause of neurodegeneration in a number of pathologies (they can, however, cause toxicity due to their sheer size). Taking Alzheimer’s disease as an example, it is now widely accepted that beta amyloid oligomers (small, soluble groups of beta amyloid proteins), as well as tau protein oligomers, are the most likely culprits for the initiation of neurotoxicity. Similarly, some studies suggest that the accumulation of oligomeric forms of alpha-synuclein and huntingtin is associated with neurotoxicity in Parkinson’s and Huntington’s disease respectively. In fact, some evidence seems to suggest that the presence of larger aggregates can protect cells from the damaging action of the above-mentioned oligomers. The proteasome, with its critical function of degrading misfolded proteins, normally ensures elimination of these proteins so they could not cause further damage to the cell. However, during neurodegeneration, something goes awry in the protein degradation mechanism. As a consequence, oligomers of misfolded proteins will escape digestion, playing a key role in pathogenesis.
Dr Smith and his collaborators showed that oligomer-mediated impairment of the proteasome is not dependent on the sequence of the misfolded protein but rather on the three-dimensional structure of the oligomer. The proteasome consists of a complex of enzymes, known as proteases, with multiple active sites that bind to proteins and degrade them rapidly and effectively inside of a protected compartment within the proteasome. Dr Smith’s studies show that when a specific type of oligomers of beta-amyloid, alpha-synuclein and huntingtin bind to the proteasome, they cause a change in its shape, denying access of the protein substrates to the degradation chamber, thereby preventing their degradation.
In-depth knowledge of the mechanism of inhibition could provide researchers worldwide with a roadmap for the rational design of proteasome activator molecules.
A common mechanism of proteasome impairment
Since these particular inhibitory oligomers have been found in many different neurodegenerative diseases, Dr Smith and collaborators suggest that there is a common mechanism of proteasome inhibition shared in these diseases. The researchers admit that other studies by different teams have already pointed out that oligomers of misfolded proteins cause inhibition of the UPS. The novelty in their particular study is that the amino acid sequence of the protein is not an important factor for its ability to inhibit the proteasome; however, the misfolding of its structure into a specific shape is critical for the development of potent proteasomal inhibition.
As protein degradation decreases and oligomers accumulate, the level of proteasome impairment increases, leading to more proteins accumulating and forming oligomers. This leads to a response that is consistent with the exponential rate of neural deterioration observed in most neurodegenerative diseases.
Pharmacological activation of the proteasome – drug discovery perspectives
Dr Smith has co-authored a comprehensive review in 2019, pointing out that proteasome inhibition is useful for treating several types of cancer. At the time when the review was written, three inhibitors were approved for use in the clinic for the treatment of blood cancers and several more inhibitors were in preclinical and clinical testing. Given that the inhibition of the proteasome can lead to the accumulation of neurotoxicity and neurodegenerative-like pathologies, Dr Smith and his colleagues argue that the pharmacological activation of the proteasome would constitute a good strategy to potentially treat neurodegenerative diseases. The activation could be achieved by the action of molecules that can induce a structural change in the entrance regions of the proteasomal binding sites – in an opposite fashion to how inhibitory oligomers do, but in this instance making the sites more accessible to protein substrates, which could rescue proteasomal function in these diseases.
To date, no drugs are known to directly activate the proteasome. However, the team hopes that the in-depth knowledge of the mechanism of inhibition could provide researchers worldwide with a roadmap for the rational design of proteasome activator molecules. These could be optimised for safety, potency and efficacy, offering a promising route for the pharmacological treatment of these devastating neurodegenerative diseases.
Personal Response
What inspired you to conduct this research?
<> Diseases like Alzheimer’s and Parkinson’s disease are becoming so pervasive in our world and touch so many people and families, and yet these diseases are fundamentally influenced by the failure of a process that we are experts at studying. This view of neurodegenerative diseases is what inspired us to dig deeper into understanding why misfolded proteins are not degraded properly in these diseases, which allows them to become toxic and cause disease. Ultimately, we thought that we could use our knowledge and skills to shed light on neurodegenerative diseases, and perhaps pave a path forward for their treatment.
Will you conduct further studies into the design of proteasome activator molecules?
<> It is a primary objective of our lab to understand the fundamental mechanisms of proteasome function, how it is impaired in neurodegenerative disease, and how we can design new molecules that could affect proteasome function in ways that might be useful to treat these devastating diseases.