The trials and tribulations in the drug discovery cycle: A particular case for Alzheimer’s disease
Alzheimer’s disease is a neurodegenerative condition that falls under the umbrella term dementia, and comprises 60-70% cases of dementia. The condition targets the elderly, with 30% of people over the age of 80 showing a loss of cognitive capabilities, which slowly affects their memory, decision-making and personality until they can no longer independently support themselves. Alzheimer’s can be a devastating disease; a manifestation of the most terrifying nightmares that haunt our collective imagination – the loss of your identity. Our art and literature are dominated by our desire to craft and cement an identity on the path of self-actualisation. Nothing is quite as scary as the inexorable loss of an identity defined by a lifetime of choices, and by an inability to remember the memories of the past or activities of the present which imbue meaning in our lives.
Alzheimer’s disease is also becoming more prevalent. The World Health Organization projects there to be 152 million dementia patients by 2050. The cost of care for Alzheimer’s patients is estimated to quintuple in the next 30 years from $226 billion to potentially $1.1 trillion. As the populations of many countries grow older, as their citizens live longer, a disease like Alzheimer’s that develops in an elderly population will only become a bigger problem.

Amyloid-β and γ-secretase enzyme
In their bid to find drug treatments against Alzheimer’s disease and on the basis of compelling genetic evidence researchers have focused on brain amyloid beta (Aβ). Aβ peptide is produced by sequential cleavage of amyloid precursor protein (APP) by beta amyloid cleavage enzyme 1 (BACE1) and gamma secretase (GS). They are peptides – molecules built from short (< 50) chains of amino acids, the building blocks of the longer proteins, The Aβ peptides come in many different shapes and sizes, the most common being Aβ38, Aβ40 and Aβ42 – the numbers denoting the number of amino acids in the peptide. The longer Aβ42 peptides are more likely to aggregate into amyloid plaques within the cerebral spinal fluid, which drive the neurocognitive symptoms that define Alzheimer’s disease.
Targeting γ-secretase becomes a very dextrous affair, as you balance the need to reduce its output for Aβ42 without disrupting other vital cellular processes.
Alzheimer’s research has identified a clear relationship between an increase in toxic Aβ42 peptides, relative to Aβ40 peptides. They’re created in an intricately complex biochemical process. Multiple enzymes cut, reshape, and add new chemicals to the amyloid precursor protein (APP), pruning it into the Aβ peptide, like a bonsai tree. Targeting this process which produces Aβ peptides has been the goal of drug development. This has yielded a variety of approaches, including targeting the γ-secretase enzyme which is primarily responsible for cutting the protein into its final lengths (Aβ42, Aβ40, or Aβ38).
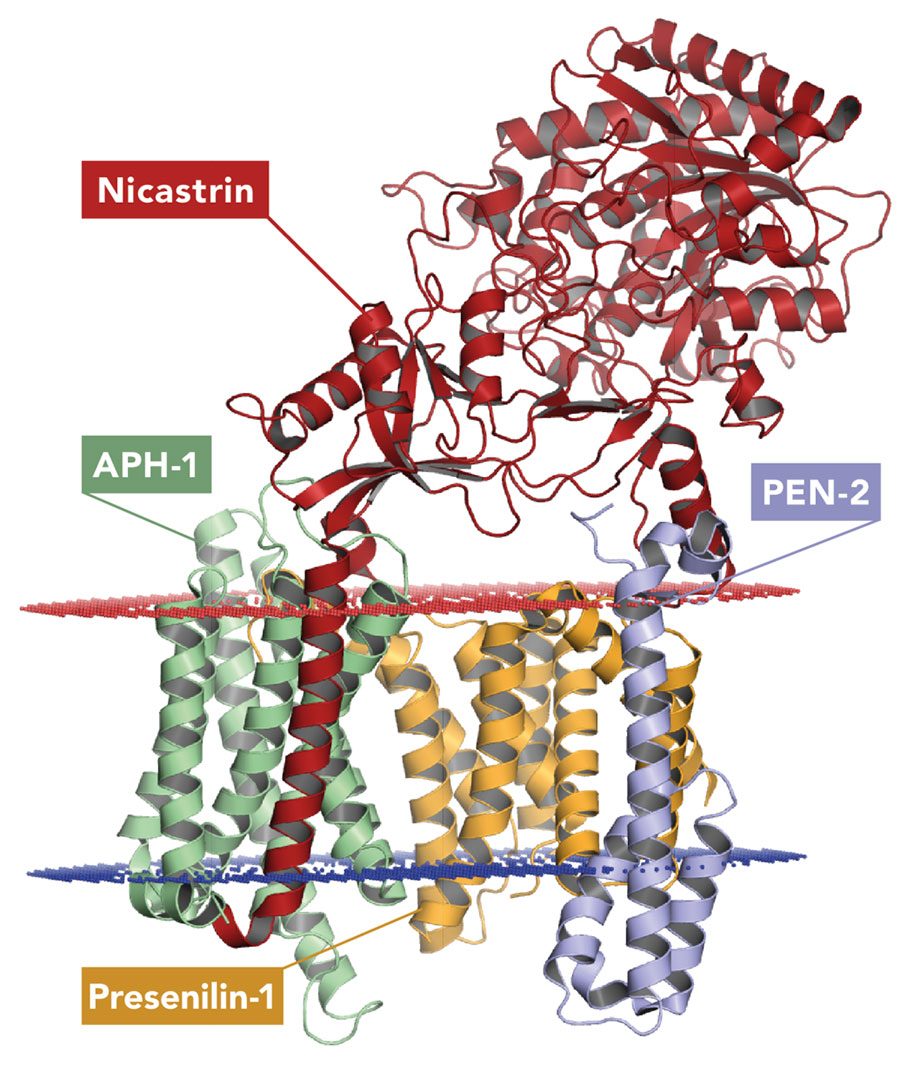
Since its discovery in 2001, researchers have targeted γ-secretase with gamma secretase modulators (GSMs). However, this is a much trickier task than it may seem. γ-secretase has its catalytic finger in many physiological pies – in other words, γ-secretase is involved in multiple chemical reactions and molecular pathways. It cleaves a broad type of protein (type-I transmembrane proteins), including Notch molecules, which are vital for cell development. It was important to observe that gamma secretase allosteric binders (“GSMs”) decrease Aβ42 and Aβ40 peptides without affecting the total amount of Aβ produced. As a result, there is an increase of shorter and beneficial Aβ38, Aβ37 peptides, offering additional therapeutic benefit for Alzheimer´s disease.
You can’t just turn it off – drugs that inhibit γ-secretase outright were abandoned after clinical trials found they were toxic. Targeting γ-secretase becomes a very dextrous affair, as you balance the need to reduce its output for Aβ42 without disrupting other vital cellular processes. This hints at the fundamental complexities involved in any effort to discover and develop new drugs.
The drug discovery cycle, but without new discoveries
The fundamental problem the drug discovery cycle poses is one of conflict, an almost contradiction in terms. The drug must simultaneously stop the process inadvertently causing the disease, but none of the other processes in the body. Drug discovery becomes a “multi-parameter” or a “multi-dimensional optimization problem”, as each candidate compound has to meet many different requirements to fulfil the desired target profile according to the disease, for its approval.
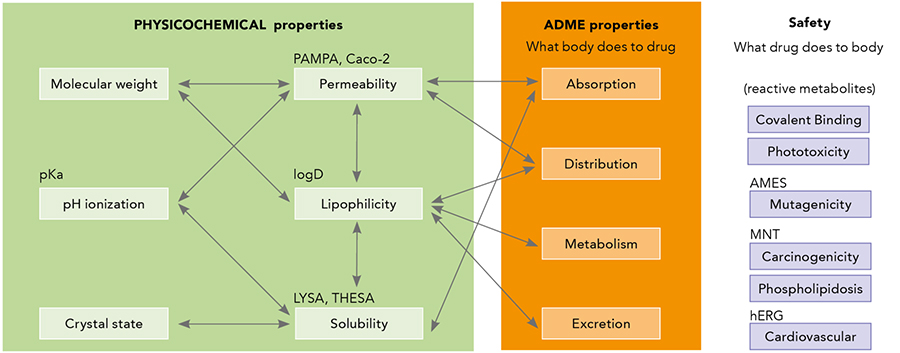
A drug candidate for Alzheimer’s disease needs to have acceptable physicochemical properties (such as its solubility and lipophilicity) and tick off the ADME list – absorption, distribution, metabolism and elimination of the drug. Researchers have to juggle this together with safety concerns by reducing the risk of toxicity eliminating observed flags, alongside the additional caveat of being able to pass through the blood-brain barrier to reach the central nervous system where the amyloid plaques form. Medicinal chemists, within a team with diverse backgrounds, need to integrate available in vitro and in vivo multi-parameters to design the next round of optimized compounds.
It becomes a see-saw of priorities; you can improve the potency of your drug, but at the expense of a long list of properties that could influence its absorption, safety, efficacy or other parameters. Juggling multiple parameters in medicinal chemistry is highly demanding and requires not only expertise but creativity and precision.
Optimization should be accomplished before going into advanced stages where safety margins are evaluated in toxicity studies, before entering human clinical trials to evaluate the risk/benefit of the drug. This is a major factor behind the stagnating research and development (R&D) scene, as the screening for ADME and low toxicity drives up costs in research, now estimated to be $1.1 billion to bring a drug to the market. Many smaller companies are locked out of drug research entirely into the Valley of Death, whilst large companies and institutes will balk at the cost of drug research, which isn’t even guaranteed to pay off.
The multi-parameter optimization problem in drug discovery (case of an Alzheimer´s drug)
A good example of the complexity of multi-parameter optimization in drug discovery can be found in the published medicinal chemistry work led by Dr Rosa María Rodríguez Sarmiento at Hoffmann La Roche Ltd. The Roche team, based on the data from biology, has long been interested in a GSM compound that can specifically alter the production of amyloid beta peptides, decreasing the proportion of Aβ42 in favour of shorter Aβ peptides that attenuate Aβ42 toxicity.
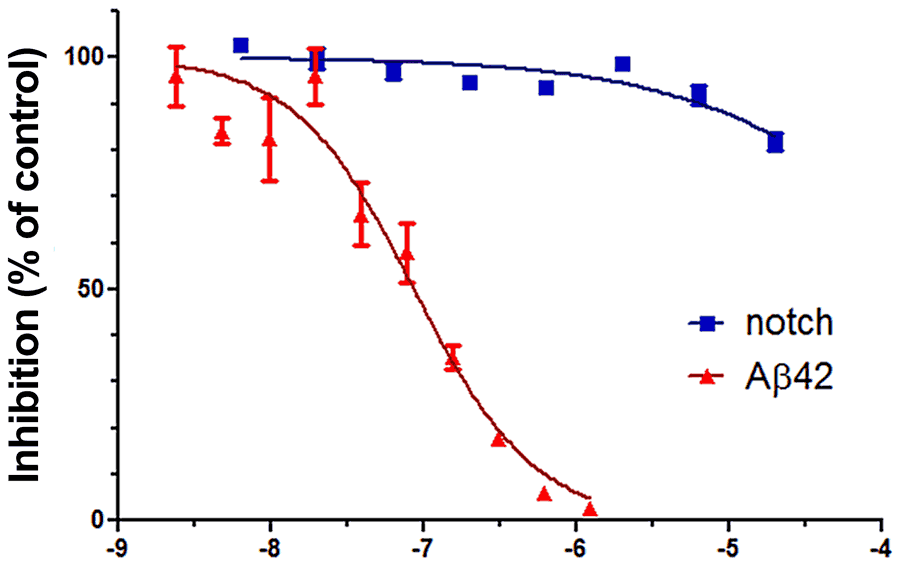
One compound which showed promise was RO6800020, as it had a strong affinity for selectively reducing the production of Aβ42, Aβ40 peptides, while increasing Aβ38, Aβ37, without interfering with Notch signalling in γ-secretase. However, it was also plagued with problems. There were multiple structural characteristics which led to toxicity, as well as a limited potency of the drug. A limited potency means, in this example, a moderate and unpredictable concentration of the drug is required to have an impact, which exacerbates existing concerns about its toxicity and makes it harder for researchers to precisely predict its effect on the patient.
Medicinal chemists, within a team with diverse backgrounds, need to integrate available in vitro and in vivo multi-parameters to design the next round of optimized compounds.
Dr Rodríguez Sarmiento saw the promise RO6800020 held and decided to look deeper into its structure, breaking it down to its core chemical constituents running through any molecules which could replace the original constituent. Through a slow, but methodical series of recombinations, drawing up new structures, Dr Rodríguez Sarmiento and her colleagues and collaborators were able to optimize its structure into RO7101556 – a much better drug in terms of properties and safety flags which was ready to be evaluated on in vivo toxicity studies as a prerequisite to go forward clinical trials.
The multi-parameter optimization solution
One important change was the central piperidine ring. This was key to increasing its potency, and its fraction unbound to acceptable levels as it meant a greater proportion of the drug molecules given in a dose weren’t attached to proteins in the plasma – therefore could do their pharmacological job of reducing Aβ42 concentrations. This also led to improving its efficacy as a drug.

In addition, the researchers realised the risk of metabolic activation of the terminal rings (“head heterocycle”) which could produce toxicity in mice liver, and replaced them with less reactive ones. In this way, they reduced toxicity by preventing covalent binding liability (CVB) to cellular macromolecules due to the ring. They also made the novel adjustment of adding a 5-substituent that prevents the drug from binding to other enzymes, specifically the PIK4CB kinase, which play an important role in triggering the immune response, preventing side effects from unselective binding to those enzymes.
However, the CF3 alkoxy is arguably the key adjustment that was made due to its ability to perfectly balance the characteristics a drug candidate needs to succeed. The Roche researchers replaced a phenyl ring with the CF3 alkoxy, which also has fluoro substituents, whilst providing many improvements through its structural differences. It can maintain the compound’s potency and selectivity towards blocking γ-secretase’s creation of amyloid beta, and Aβ42, and balances this with the traits that make it a viable preclinical drug candidate for further exploration in dose range toxicity before becoming a clinical candidate.
OCF3 absorbs significantly less ultraviolet A waves, thus reducing toxicity from UV radiation. Alongside this, the less aromatic structure of OCF3 makes it less lipophilic and more soluble, helping it flow in the bloodstream and reducing the chance of the drug binding to plasma proteins. Finally, the CF3 alkoxy is slowly metabolised so it maintains a good pharmacokinetic (ADME) profile and a low P-glycoprotein (Pgp) mediated efflux, ensuring it can pass through the blood-brain barrier and reach the amyloid plaques.
It’s undeniable that the complex balancing act makes drug discovery and development a time-consuming and resource intensive process. For diseases like Alzheimer’s, there is a pressing need for a detailed and thorough approach which can investigate an abundance of potential changes to meet the many requirements for drug treatments. The only solution to a multi-faceted problem is a multi-faceted solution.
Personal Response
Do there need to be any changes to practice within research science or funding to overcome the challenges in drug discovery? If so, what do you think should change?
Where do the specific ideas for changing or replacing chemicals/molecules in a compound come from? (for example, changing the phenyl ring to the bioisosteric CF3 alkoxy ring?)